
Carbonyl Group Reactions
- Page ID
- 1175

The metal hydride reductions and organometallic additions to aldehydes and ketones, described above, both decrease the carbonyl carbon's oxidation state, and may be classified as reductions. As noted, they proceed by attack of a strong nucleophilic species at the electrophilic carbon. Other useful reductions of carbonyl compounds, either to alcohols or to hydrocarbons, may take place by different mechanisms. For example, hydrogenation (Pt, Pd, Ni or Ru catalysts), reaction with diborane, and reduction by lithium, sodium or potassium in hydroxylic or amine solvents have all been reported to convert carbonyl compounds into alcohols. However, the complex metal hydrides are generally preferred for such transformations because they give cleaner products in high yield.
Aldehydes and ketones may also be reduced by hydride transfer from alkoxide salts.
The reductive conversion of a carbonyl group to a methylene group requires complete removal of the oxygen, and is called deoxygenation. In the shorthand equation shown here the [H] symbol refers to unspecified reduction conditions which effect the desired change. Three very different methods of accomplishing this transformation will be described here.
R2C=O + [H]  R2CH2 + H2O
R2CH2 + H2O
Wolff-Kishner Reduction
Reaction of an aldehyde or ketone with excess hydrazine generates a hydrazone derivative, which on heating with base gives the corresponding hydrocarbon. A high-boiling hydroxylic solvent, such as diethylene glycol, is commonly used to achieve the temperatures needed. The following diagram shows how this reduction may be used to convert cyclopentanone to cyclopentane. A second example, in which an aldehyde is similarly reduced to a methyl group, also illustrates again the use of an acetal protective group. The mechanism of this useful transformation involves tautomerization of the initially formed hydrazone to an azo isomer, and will be displayed on pressing the "Show Mechanism" button. The strongly basic conditions used in this reaction preclude its application to base sensitive compounds.
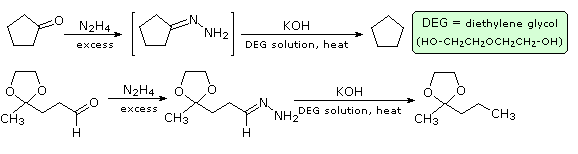
Figure 1A: Wolff-Kischner reduction of cyclopentanone
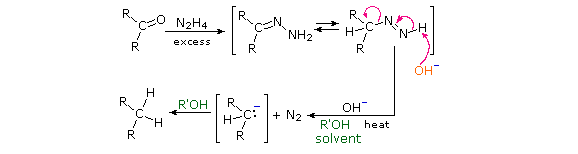
Figure 1B: Wolff-Kischner reudction of cyclopentanone
Clemmensen Reduction
This alternative reduction involves heating a carbonyl compound with finely divided, amalgamated zinc in a hydroxylic solvent (often an aqueous mixture) containing a mineral acid such as HCl. The mercury alloyed with the zinc does not participate in the reaction, it serves only to provide a clean active metal surface. The first example below shows a common application of this reduction, the conversion of a Friedel-Crafts acylation product to an alkyl side-chain. The second example illustrates the lability of functional substituents alpha to the carbonyl group. Substituents such as hydroxyl, alkoxyl & halogens are reduced first, the resulting unsubstituted aldehyde or ketone is then reduced to the parent hydrocarbon.
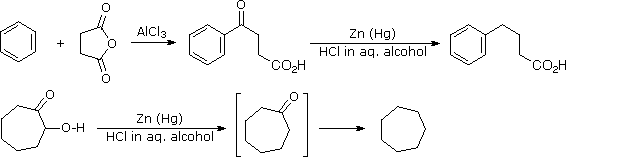
Figure 2A: Clemmensen reduction
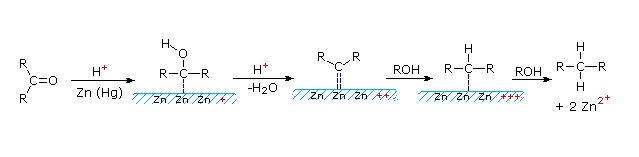
Figure 2B: Clemmensen reduction Mechanism
Hydrogenolysis of Thioacetals
In contrast to the previous two procedures, this method of carbonyl deoxygenation requires two separate steps. It does, however, avoid treatment with strong base or acid. The first step is to convert the aldehyde or ketone into a thioacetal: These derivatives may be isolated and purified before continuing the reduction. The second step involves refluxing an acetone solution of the thioacetal over a reactive nickel catalyst, called Raney Nickel. All carbon-sulfur bonds undergo hydrogenolysis (the C–S bonds are broken by addition of hydrogen). In the following example, 1,2-ethanedithiol is used for preparing the thioacetal intermediate, because of the high yield this reactant usually affords. The bicyclic compound shown here has two carbonyl groups, one of which is sterically hindered (circled in orange). Consequently, a mono-thioacetal is easily prepared from the less-hindered ketone, and this is reduced without changing the remaining carbonyl function.
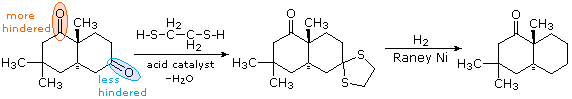
Figure 3: Raney Nickel hydrogenolysis of thioacetals
Oxidation
The carbon atom of a carbonyl group has a relatively high oxidation state. This is reflected in the fact that most of the reactions described thus far either cause no change in the oxidation state (e.g. acetal and imine formation) or effect a reduction (e.g. organometallic additions and deoxygenations). The most common and characteristic oxidation reaction is the conversion of aldehydes to carboxylic acids. In the shorthand equation shown here the [O] symbol refers to unspecified oxidation conditions which effect the desired change. Several different methods of accomplishing this transformation will be described here.
RCH=O + [O]  RC(OH)=O
RC(OH)=O
In discussing the oxidations of 1º and 2º-alcohols, we noted that Jones' reagent (aqueous chromic acid) converts aldehydes to carboxylic acids, presumably via the hydrate. Other reagents, among them aqueous potassium permanganate and dilute bromine, effect the same transformation. Even the oxygen in air will slowly oxidize aldehydes to acids or peracids, most likely by a radical mechanism. Useful tests for aldehydes, Tollens' test, Benedict's test & Fehling's test, take advantage of this ease of oxidation by using Ag(+) and Cu(2+) as oxidizing agents (oxidants).
RCH=O + 2 [Ag(+) OH(–)]  RC(OH)=O + 2 Ag (metallic mirror) + H2O
RC(OH)=O + 2 Ag (metallic mirror) + H2O
When silver cation is the oxidant, as in the above equation, it is reduced to metallic silver in the course of the reaction, and this deposits as a beautiful mirror on the inner surface of the reaction vessel. The Fehling and Benedict tests use cupric cation as the oxidant. This deep blue reagent is reduced to cuprous oxide, which precipitates as a red to yellow solid. All these cation oxidations must be conducted under alkaline conditions. To avoid precipitation of the insoluble metal hydroxides, the cations must be stabilized as complexed ions. Silver is used as its ammonia complex, Ag(NH3)2(+), and cupric ions are used as citrate or tartrate complexes.
Saturated ketones are generally inert to oxidation conditions that convert aldehydes to carboxylic acids. Nevertheless, under vigorous acid-catalyzed oxidations with nitric or chromic acids ketones may undergo carbon-carbon bond cleavage at the carbonyl group. The reason for the vulnerability of the alpha-carbon bond will become apparent in the following section.
Contributors
- William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry