
II. Oximes
- Page ID
- 24553
A. Addition Reactions
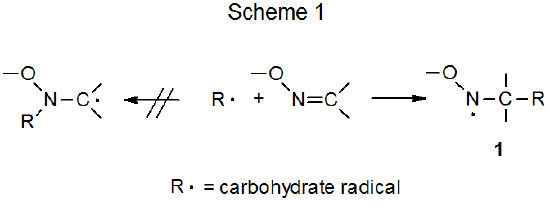
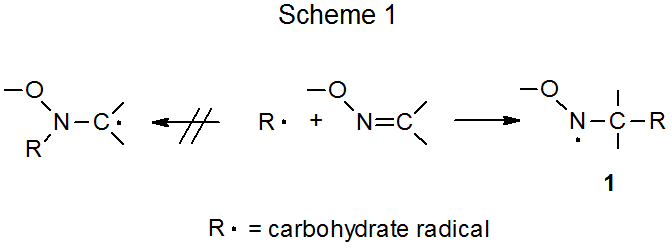
A carbon-centered radical adds preferentially to the carbon atom in the carbon–nitrogen double bond of an oxime ether (Scheme 1). The regioselectivity of this reaction is consistent with a radical adding to the oxime ether to produce the more stable of the two possible adduct radicals; that is, the radical stabilized by an oxygen atom attached to the radical center. Examples of this type of reaction are found in the addition of a pyranos-1-yl radical to O‑benzylformaldoxime (eq 1),3 and reaction of a nucleoside radical centered at C-3' to a nucleoside-containing oxime ether (eq 2).4,5
.png?revision=1&size=bestfit&width=410&height=182)
.png?revision=1&size=bestfit&width=430&height=176)
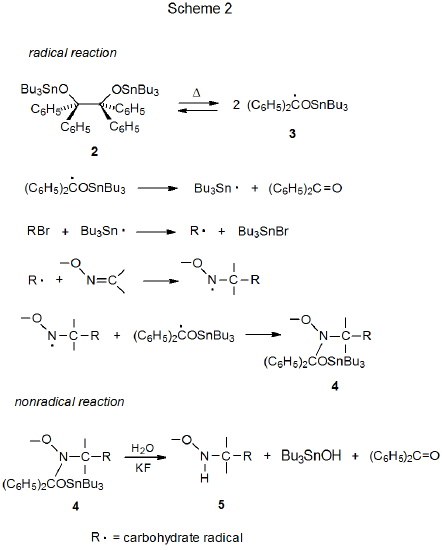
The carbon-centered radicals that add to oxime ethers typically are formed from the corresponding iodides and bromides by reaction with Bu3Sn·. Because simple reduction offers significant competition to radical addition, Bu3SnH is not the best source for the tin-centered radicals participating in these reactions. Addition is more successful when Bu3Sn· is generated from bis(tri-n-butyltin)benzpinacolate (2), a compound that produces Bu3Sn· but suppresses simple reduction by not introducing an effective hydrogen-atom transfer into the reaction mixture (Scheme 2).3 In the absence of such a donor the radical phase of this reaction is thought to produce the tin-containing compound 4, which then is hydrolyzed to the benzyloxyamine 5 in a nonradical reaction (Scheme 2).
B. Cyclization Reactions
1. Regioselectivity
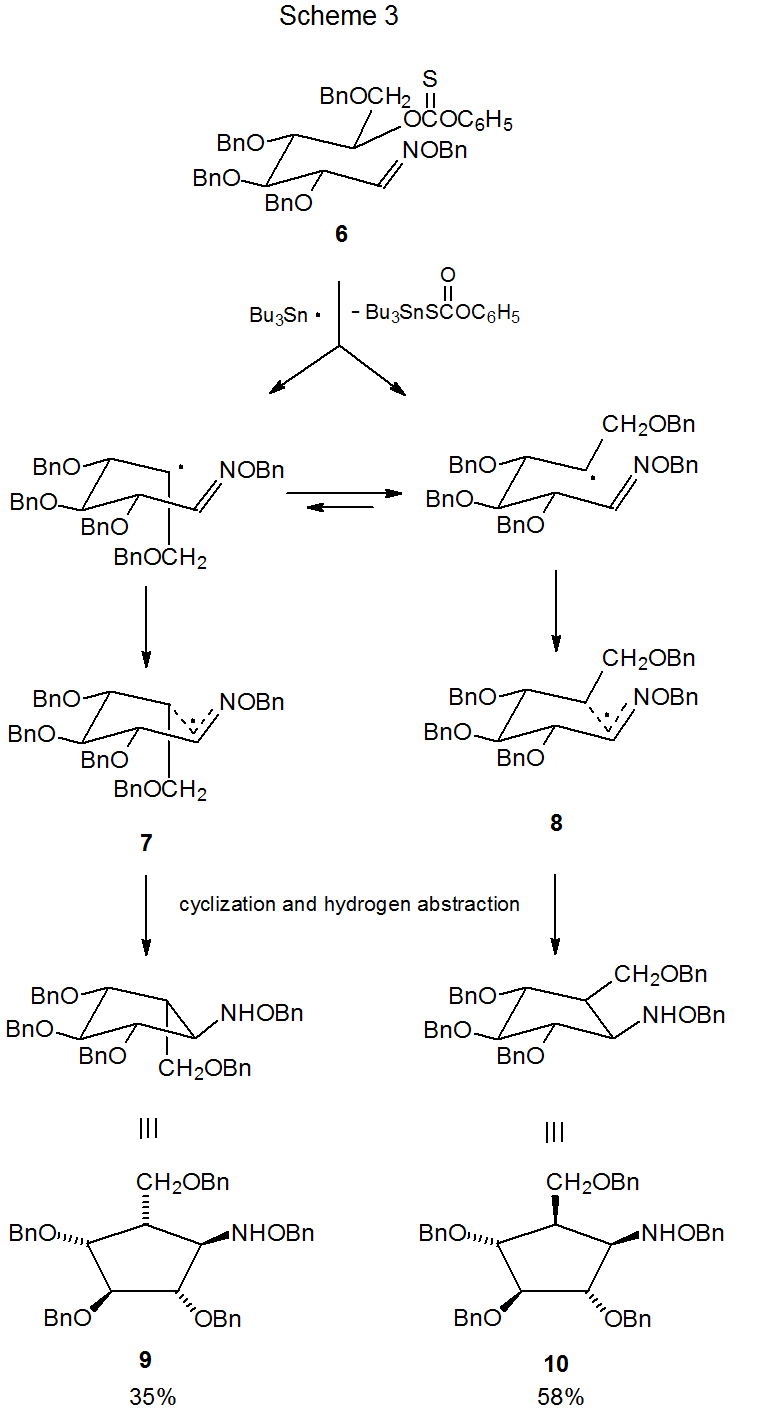
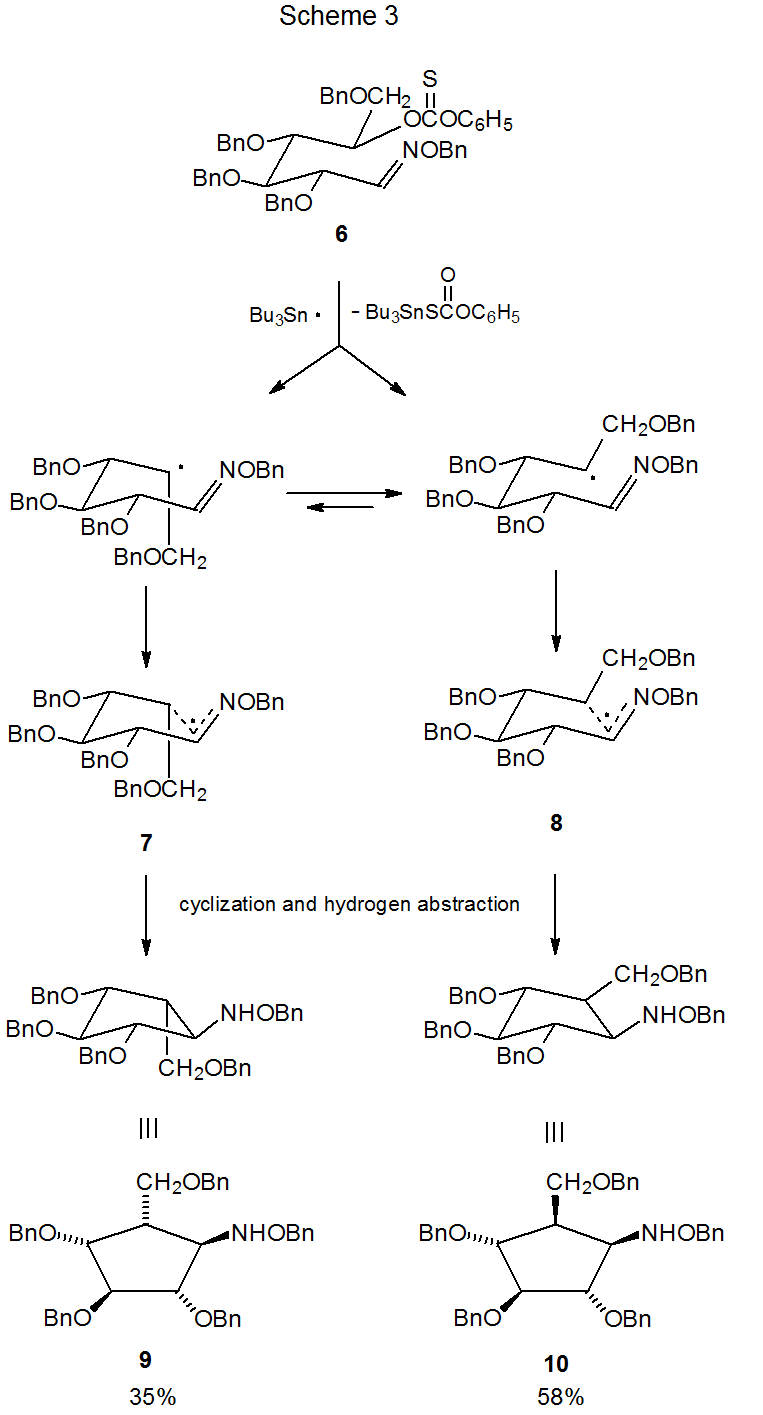
An example of regioselective radical cyclization involving an oxime ether is shown in Scheme 3.6 One factor important in determining regioselectivity in this type of reaction is the energetically favored addition of a radical to the carbon atom of the carbon–nitrogen double bond (Scheme 1). Also of importance is the preference during radical cyclization of oxime ethers for forming five-membered rather than six-membered rings when both are possible and six-membered rather than seven-membered rings when either one could be formed.7
{kind=link}
2. Stereoselectivity
Radical cyclization converts open-chain oxime ethers into highly substituted, alkoxyamino cyclopentanes6,8–14 and cyclohexanes.7,15–17 As is often the case in radical cyclization reactions, stereoselectivity can be understood, if one assumes that the lowest energy transition state is a chair-like structure that maximizes the number of pseudoequatorial substituents. The reaction shown in Scheme 3, for example, generates compounds with two new chiral centers but produces only two (9 and 10) of the four possible stereoisomers.6 These two isomers arise from the chair-like transition states 7 and 8, respectively. The transition state 8, leading to the major product 10 has the greater number of pseudoequatorial substituents.
In light of the reaction of the oxime ether 6 (Scheme 3) one might expect the structurally similar oxime ether 11 (eq 3) also to produce only the two stereoisomers 14 and 15, that is, the products expected from reaction via chair-like transition states. Actually, 11 produces all four possible stereoisomers (eq 3), and neither 14 nor 15 is the major product.11 The major product (12) arises from reaction involving the boat-like transition state 16 (Scheme 4). Similar amounts of energy often are required to reach chair-like and boat-like transition states in radical cyclization reactions (Section IV.A in Chapter 11 of Volume I); consequently, differences in structure near the reactive centers can affect product distribution significantly.
.png?revision=1&size=bestfit&width=415&height=350)
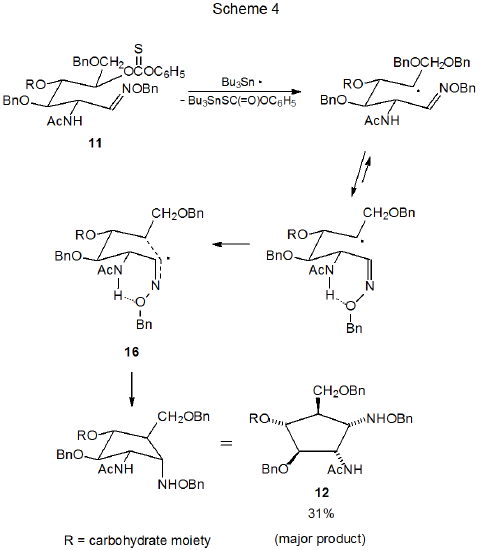
One difference between the substrates 6 and 11 is that 6 has a benzyloxy group at C-2 (Scheme 3), and 11 has an acetamido group at this position (Scheme 4). The possibility exists that hydrogen bonding involving the acetamido group stabilizes the transition state 16 to the point that the reaction pathway containing this boat-like structure (16) becomes the lowest energy one (Scheme 4).
{kind=link}
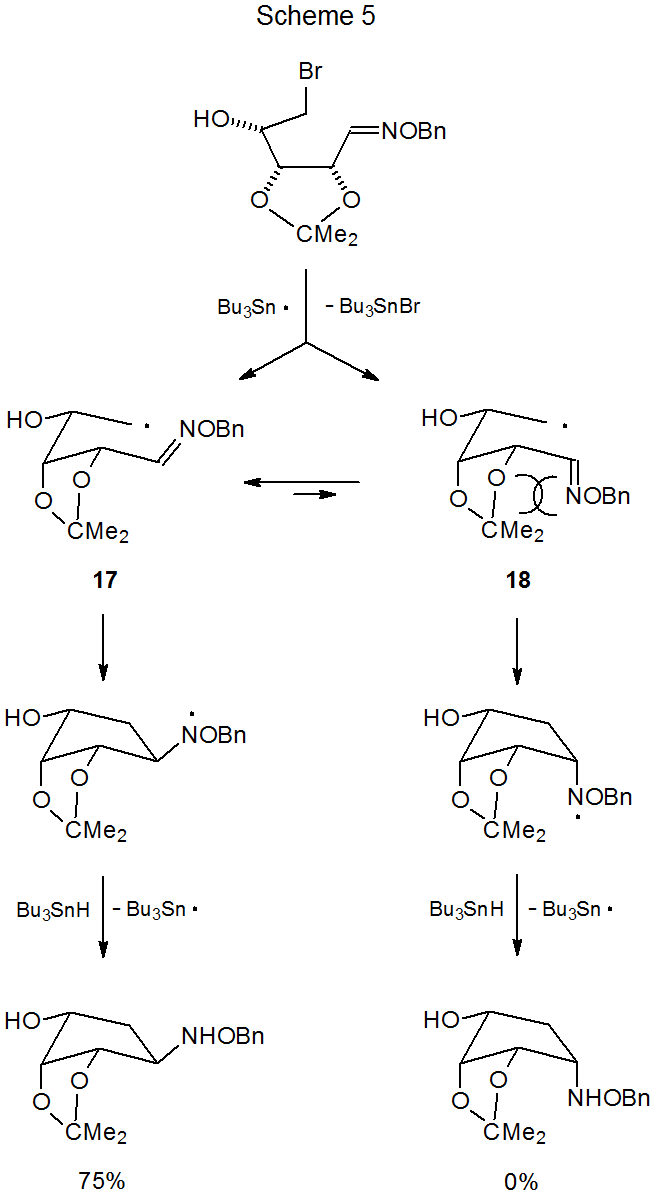
An O-isopropylidene group that is close to the reactive centers in a cyclization reaction has a pronounced effect of stereoselectivity.17–24 This effect is illustrated by the reaction shown in Scheme 5, where the sole product has the NHOBn substituent on the side of the newly formed ring that is opposite to the O-isopropylidene group.18 In this reaction the chair-like conformation (17) of the intermediate radical is greatly favored over its boat-like counterpart (18) due, in large measure, to destabilizing steric interactions that involve the O-isopropylidene group.
3. Radical Forming Reactions
Carbohydrate radicals that add internally to carbon–nitrogen double bonds come from a variety of sources. Such radicals often form by reaction of O-thiocarbonyl compounds6,8–11,20,21,23 or bromides15–19 with tri-n-butyltin hydride. Reactions of Bu3SnH with carbonyl compounds12 and dithioacetals13 represent two less common ways for generating radicals that then undergo ring formation. The necessary radicals also can be formed by electron-transfer reaction.
4. Preserving the Double Bond
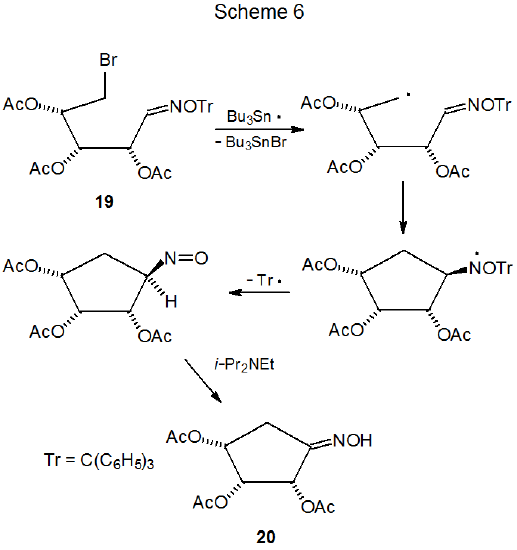
Normally, the double bond in an oxime ether is converted into a single bond during ring formation, but when an O-trityl oxime undergoes cyclization, the carbon–nitrogen double bond is preserved in the product. A proposed mechanism for this type of reaction is shown in Scheme 6, and the preferred reagents, all of which are critical to the success of the reaction, are given in eq 4.24 Slow addition of Bu3SnH and the initiator ABC also are necessary for synthetically useful reaction. The persistent radical (C6H5)3C· hinders chain propagation by its slow reaction with Bu3SnH; consequently, to avoid buildup of (C6H5)3C·, diphenyl diselenide, a convenient precursor for the very reactive hydrogen-atom transfer C6H5SeH, can be added to the reaction mixture. This addition replaces a slow reaction (eq 5) with two rapid ones (eq 6 and eq 7).24 Although the carbohydrates that have been studied thus far give good product yields without addition of diphenyl diselenide, other compounds do not; consequently, the selenide inclusion in the reaction mixture is now part of the recommended procedure.
.png?revision=1&size=bestfit&width=225&height=128)
.png?revision=1&size=bestfit&width=330&height=21)
.png?revision=1&size=bestfit&width=385&height=22)
.png?revision=1&size=bestfit&width=340&height=21)
5. Enhanced Reactivity of Vinylic Radicals
Attempted cyclization of the radical generated from the bromide 21 produces only the simple-reduction product 22 (eq 8), but reaction of the related vinylic bromide 23 gives both a cyclic product and a simple-reduction product (eq 9).25 One factor that may contribute to the difference in ability of compounds 21 and 23 to undergo radical cyclization is the considerable reactivity of vinylic radicals. This reactivity may be sufficient to cause new ring formation during reaction of 23.
.png?revision=1&size=bestfit&width=360&height=144)
.png?revision=1&size=bestfit&width=360&height=251)
In addition to halogen-atom abstraction, vinylic radicals also form by reaction of a tin-centered radical with a compound containing a carbon–carbon triple bond.26–30 When a radical formed in this way adds internally to a carbon–nitrogen double bond, it produces a cyclic, tin-containing product (eq 10).26 The triple bond in such reactions usually is a terminal one, but internal bonds, particularly those in conjugation with radical-stabilizing groups, also undergo reaction.30
.png?revision=1&size=bestfit&width=395&height=116)
C. Electron-Transfer Reaction
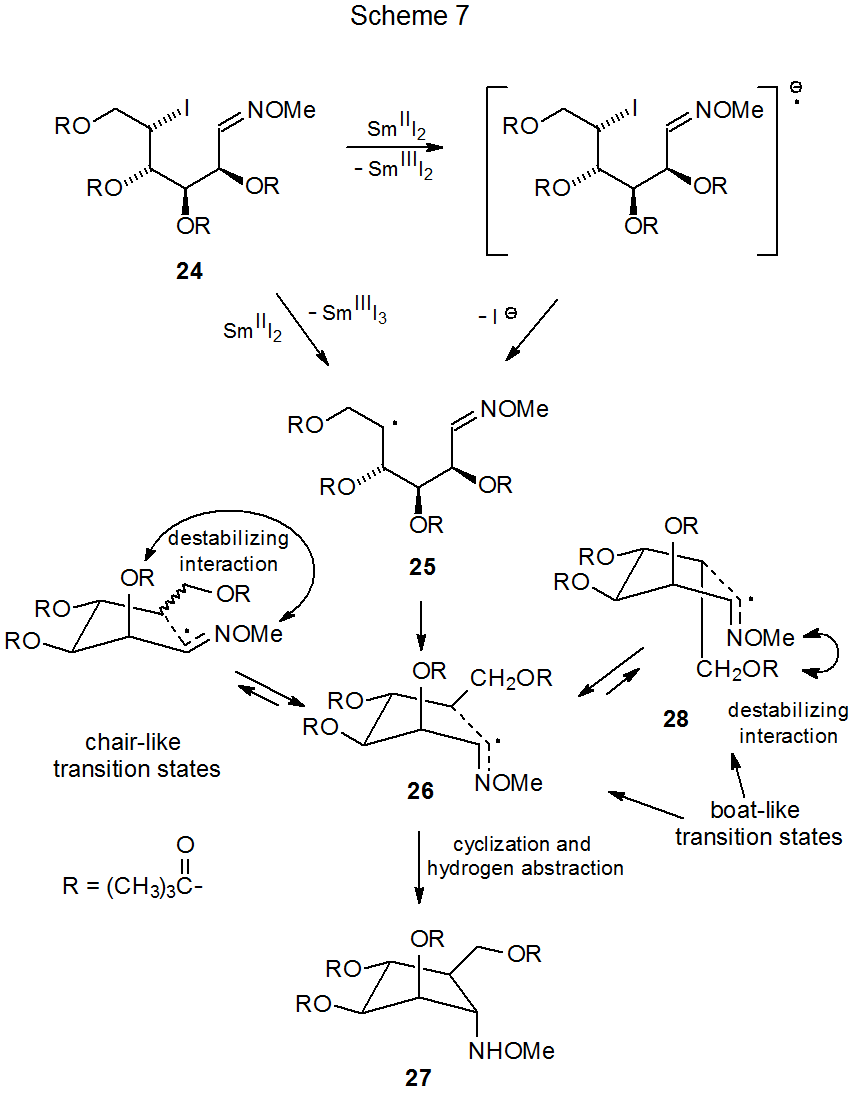
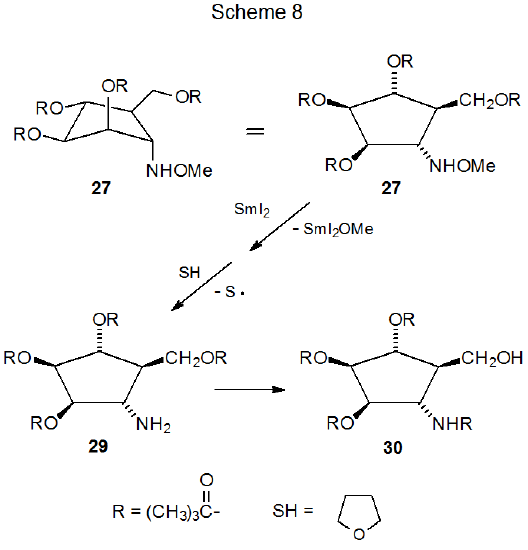
Electron transfer provides another means for generating a ring-forming radical from an oxime ether. The electron donor in such reactions typically is samarium(II) iodide and the electron acceptor often is an oxime ether containing a halogen atom. A typical reaction is shown in Scheme 7,31 where dissociative electron transfer involving the carbon–iodine bond in 24 leads to formation of the carbon-centered radical 25. Cyclization and hydrogen-atom abstraction complete this reaction by forming the cyclopentylamine derivative 27 in a process that passes through the boat-like transition state 26. Although chair-like transition states typically are favored in cyclization reactions, in this case chair-like structures have destabilizing interaction between C-1 and C-2 substituents; furthermore, the other boat-like transition state (28) has reduced stability due to the interaction between the C-1 and C-6 substituents (Scheme 7). If excess SmI2 is present during reaction, dissociative electron transfer breaks the N–O bond in 27 to produce, after hydrogen-atom abstraction, the corresponding cyclopentylamine 29 (Scheme 8). Nonradical migration of the acyl group from oxygen to nitrogen then forms the final product (30).31 (Samarium(II) iodide also cleaves N–O bonds that are not formed from reaction of oxime ethers.32)
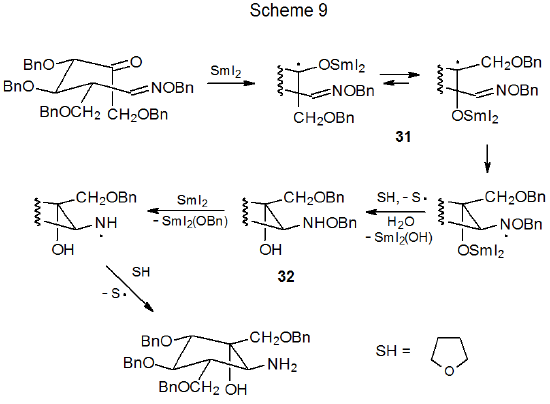
Electron transfer to an aldehydo or keto group in a oxime ether also can be the initial step in ring formation.33–38 When SmI2 is the electron-donor, the first reactive intermediate is a samarium ketyl formed by one-electron reduction of the carbonyl group.1 In the reaction shown in Scheme 9, the carbon atom on which the radical in the samarium ketyl 31 is centered adds internally to the carbon–nitrogen double bond leading to the cyclic product 32. When excess SmI2 is present, electron transfer to 32 causes N–O bond cleavage and ultimate replacement of the O-benzyl group attached to nitrogen with a hydrogen atom (Scheme 9).36 Similar cyclization has been observed when the tri-n-butyltin radical reacts with a oxime ether containing an aldehydo or keto group.39
D. Oxime Esters as Radical Sources
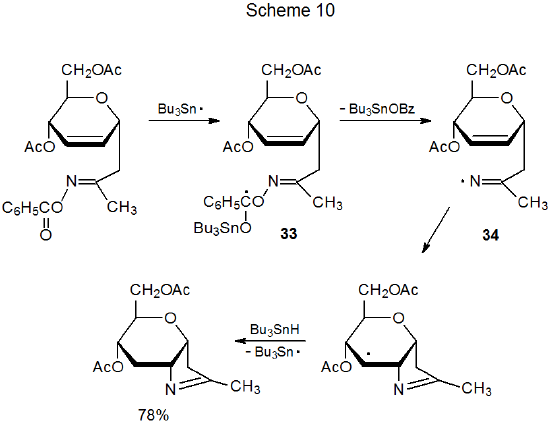
The signature event of the cyclization reactions discussed thus far is a carbon-centered radical adding to a carbon–nitrogen double bond in an oxime ether. The majority of ring-forming reactions involving oxime derivatives take place in this way, but a change occurs in the reaction shown in Scheme 10, where the adding radical (34) is generated from an O-benzoyloxime.40 In general, adding Bu3Sn· to an O-benzoyl group is an inauspicious beginning for a radical reaction because such addition reverses rapidly, nearly always before any other reaction can occur. Further reaction is possible in this case (Scheme 10) due primarily to rapid fragmentation of the weak N‑O bond in the initially formed radical 33.40
E. Oxime Synthesis
Oximes can be the products of radical reaction.41,42 When the cobalt-containing, D‑glucopyranosyl derivative 35 is photolyzed, the radical produced adds to a molecule of nitric oxide to give a nitroso compound. This compound then tautomerizes to form the corresponding oxime 36 (eq 11).41
.png?revision=1&size=bestfit&width=420&height=213)