16.8: Electrochemical Corrosion
- Page ID
- 265

Make sure you thoroughly understand the following essential ideas. It is especially important that you know the precise meanings of all the highlighted terms in the context of this topic.
- Electrochemical corrosion of metals occurs when electrons from atoms at the surface of the metal are transferred to a suitable electron acceptor or depolarizer. Water must be present to serve as a medium for the transport of ions.
- The most common depolarizers are oxygen, acids, and the cations of less active metals.
- Because the electrons flow through the metallic object itself, the anodic and cathodic regions (the two halves of the electrochemical cell) can be at widely separated locations.
- Anodic regions tend to develop at locations where the metal is stressed or is protected from oxygen.
- Contact with a different kind of metal, either direct or indirect, can lead to corrosion of the more active one.
- Corrosion of steel can be inhibited by galvanizing, that is, by coating it with zinc, a more active metal whose dissolution leaves a negative charge on the metal which inhibits the further dissolution of Fe2+.
- Cathodic protection using an external voltage source is widely used to protect underground structures such as tanks, pipelines and piers. The source can be a sacrificial anode of zinc or aluminum, or a line-operated or photovoltaic power supply.
Corrosion can be defined as the deterioration of materials by chemical processes. Of these, the most important by far is electrochemical corrosion of metals, in which the oxidation process M → M+ + e– is facilitated by the presence of a suitable electron acceptor, sometimes referred to in corrosion science as a depolarizer.
In a sense, corrosion can be viewed as the spontaneous return of metals to their ores; the huge quantities of energy that were consumed in mining, refining, and manufacturing metals into useful objects is dissipated by a variety of different routes. The economic aspects of corrosion are far greater than most people realize; the estimated cost of corrosion in the U.S. alone was $276 billion per year. Of this, about $121 billion was spent to control corrosion, leaving the difference of $155 billion as the net loss to the economy. Utilities, especially drinking water and sewer systems, suffer the largest economic impact, with motor vehicles and transportation being a close second.
Corrosion Cells and Reactions
The special characteristic of most corrosion processes is that the oxidation and reduction steps occur at separate locations on the metal. This is possible because metals are conductive, so the electrons can flow through the metal from the anodic to the cathodic regions (Figure \(\PageIndex{1}\)). The presence of water is necessary in order to transport ions to and from the metal, but a thin film of adsorbed moisture can be sufficient.
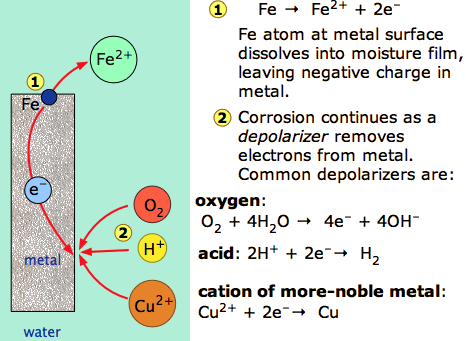
A corrosion system can be regarded as a short-circuited electrochemical cell in which the anodic process is something like
\[\ce{Fe(s) \rightarrow Fe^{2+}(aq) + 2 e^{-}} \label{1.7.1}\]
and the cathodic steps may invove the reduction of oxygen gas
\[ \ce{O_2} + \ce{2 H_2O} + \ce{4e^{-}} \rightarrow \ce{4 OH^{-}} \label{1.7.2}\]
or the reduction of protons
\[ \ce{H^{+} + e^{-}} \rightarrow \ce{1/2 H2(g)} \label{1.7.2b} \]
or the reduction of a metal ion
\[\ce{M^{2+} + 2 e^{–}} \rightarrow \ce{M(s)} \label{1.7.2c}\]
where \(\ce{M}\) is a metal.
Which parts of the metal serve as anodes and cathodes can depend on many factors, as can be seen from the irregular corrosion patterns that are commonly observed. Atoms in regions that have undergone stress, as might be produced by forming or machining, often tend to have higher free energies, and thus tend to become anodic.
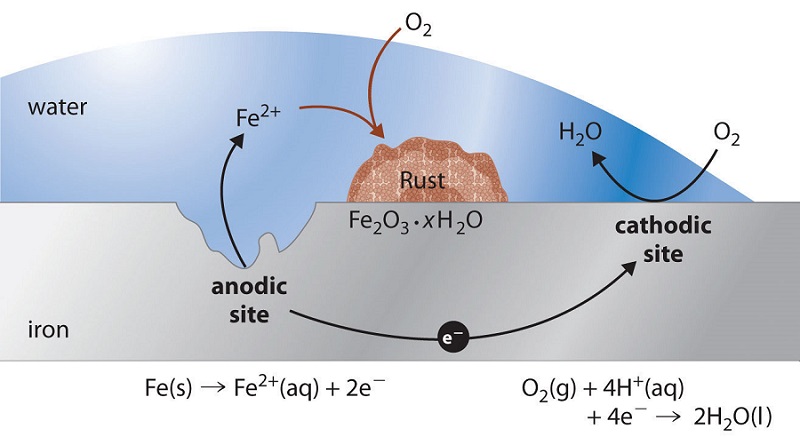
If one part of a metallic object is protected from the atmosphere so that there is insufficient \(\ce{O2}\) to build or maintain the oxide film, this "protected" region will often be the site at which corrosion is most active. The fact that such sites are usually hidden from view accounts for much of the difficulty in detecting and controlling corrosion.
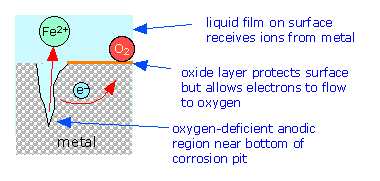
In contrast to anodic sites, which tend to be localized to specific regions of the surface, the cathodic part of the process can occur almost anywhere. Because metallic oxides are usually semiconductors, most oxide coatings do not inhibit the flow of electrons to the surface, so almost any region that is exposed to \(\ce{O2}\) or to some other electron acceptor can act as a cathode. The tendency of oxygen-deprived locations to become anodic is the cause of many commonly-observed patterns of corrosion.
Rusted-out Cars and Bathroom Stains
Anyone who has owned an older car has seen corrosion occur at joints between body parts and under paint films. You will also have noticed that once corrosion starts, it tends to feed on itself. One reason for this is that one of the products of the O2 reduction reaction is hydroxide ion. The high pH produced in these cathodic regions tends to destroy the protective oxide film, and may even soften or weaken paint films, so that these sites can become anodic. The greater supply of electrons promotes more intense cathodic action, which spawns even more anodic sites, and so on.

A very common cause of corrosion is having two dissimilar metals in contact, as might occur near a fastener or at a weld joint. Moisture collects at the junction point, acting as an electrolyte and forming a cell in which the two metals serve as electrodes. Moisture and conductive salts on the outside surfaces provide an external conductive path, effectively short-circuiting the cell and producing very rapid corrosion; this is why cars rust out so quickly in places where salt is placed on roads to melt ice.
Dissimilar-metal corrosion can occur even if the two metals are not initially in direct contact. For example, in homes where copper tubing is used for plumbing, there is always a small amount of dissolved \(\ce{Cu^{2+}}\) in the water. When this water encounters steel piping or a chrome-plated bathroom sink drain, the more-noble copper will plate out on the other metal, producing a new metals-in-contact corrosion cell. In the case of chrome bathroom sink fittings, this leads to the formation of \(\ce{Cr^{3+}}\) salts which precipitate as greenish stains.
Control of Corrosion
Since both the cathodic and anodic steps must take place for corrosion to occur, prevention of either one will stop corrosion. The most obvious strategy is to stop both processes by coating the object with a paint or other protective coating. Even if this is done, there are likely to be places where the coating is broken or does not penetrate, particularly if there are holes or screw threads. A more sophisticated approach is to apply a slight negative charge to the metal, thus making it more difficult for the reaction to take place:
\[\ce{M -> M^{2+} + 2 e^{-}}.\]
Protection Method 1: Sacrificial Coatings
One way of supplying this negative charge is to apply a coating of a more active metal. Thus a very common way of protecting steel from corrosion is to coat it with a thin layer of zinc; this process is known as galvanizing.The zinc coating, being less noble than iron, tends to corrode selectively. Dissolution of this sacrificial coating leaves behind electrons which concentrate in the iron, making it cathodic and thus inhibiting its dissolution.

The effect of plating iron with a less active metal provides an interesting contrast. The common tin-plated can (on the right) is a good example. As long as the tin coating remains intact, all is well, but exposure of even a tiny part of the underlying iron to the moist atmosphere initiates corrosion. The electrons released from the iron flow into the tin, making the iron more anodic so now the tin is actively promoting corrosion of the iron! You have probably observed how tin cans disintegrate very rapidly when left outdoors.
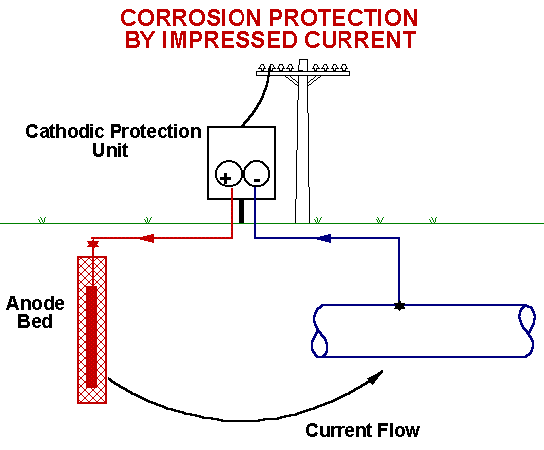
Protection Method 2: Cathodic Protection
A more sophisticated strategy is to maintain a continual negative electrical charge on a metal, so that its dissolution as positive ions is inhibited. Since the entire surface is forced into the cathodic condition, this method is known as cathodic protection. The source of electrons can be an external direct current power supply (commonly used to protect oil pipelines and other buried structures), or it can be the corrosion of another, more active metal such as a piece of zinc or aluminum buried in the ground nearby, as is shown in the illustration of the buried propane storage tank below.