
Carboxylic Derivatives - Reactions (Acyl Group Substitution and Mechanism)
- Page ID
- 1217

Acyl Group Substitution
This is probably the single most important reaction of carboxylic acid derivatives. The overall transformation is defined by the following equation, and may be classified either as nucleophilic substitution at an acyl group or as acylation of a nucleophile. For certain nucleophilic reagents the reaction may assume other names as well. If Nuc-H is water the reaction is often called hydrolysis, if Nuc–H is an alcohol the reaction is called alcoholysis, and for ammonia and amines it is called aminolysis.
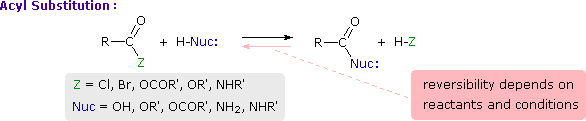
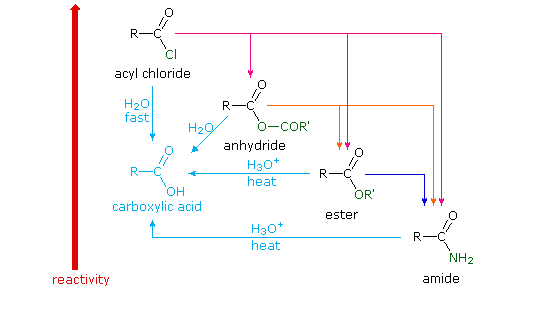
Different carboxylic acid derivatives have very different reactivities, acyl chlorides and bromides being the most reactive and amides the least reactive, as noted in the following qualitatively ordered list. The change in reactivity is dramatic. In homogeneous solvent systems, reaction of acyl chlorides with water occurs rapidly, and does not require heating or catalysts. Amides, on the other hand, react with water only in the presence of strong acid or base catalysts and external heating.
Reactivity: acyl halides > anhydrides >> esters ≈ acids >> amides
Because of these differences, the conversion of one type of acid derivative into another is generally restricted to those outlined in the following diagram. Methods for converting carboxylic acids into these derivatives were shown in a previous section, but the amide and anhydride preparations were not general and required strong heating. A better and more general anhydride synthesis can be achieved from acyl chlorides, and amides are easily made from any of the more reactive derivatives. Specific examples of these conversions will be displayed by clicking on the product formula. The carboxylic acids themselves are not an essential part of this diagram, although all the derivatives shown can be hydrolyzed to the carboxylic acid state (light blue formulas and reaction arrows). Base catalyzed hydrolysis produces carboxylate salts.
Before proceeding further, it is important to review the general mechanism by means of which all these acyl transfer or acylation reactions take place. Indeed, an alert reader may well be puzzled by the facility of these nucleophilic substitution reactions. After all, it was previously noted that halogens bonded to sp2 or sp hybridized carbon atoms do not usually undergo substitution reactions with nucleophilic reagents. Furthermore, such substitution reactions of alcohols and ethers are rare, except in the presence of strong mineral acids. Clearly, the mechanism by which acylation reactions occur must be different from the SN1 and SN2 procedures described earlier.
In any substitution reaction two things must happen. The bond from the substrate to the leaving group must be broken, and a bond to the replacement group must be formed. The timing of these events may vary with the reacting system. In nucleophilic substitution reactions of alkyl compounds examples of bond-breaking preceding bond-making (the SN1 mechanism), and of bond-breaking and bond-making occurring simultaneously (the SN2 mechanism) were observed. On the other hand, for most cases of electrophilic aromatic substitution bond-making preceded bond-breaking.
As illustrated in the following diagram, acylation reactions generally take place by an addition-elimination process in which a nucleophilic reactant bonds to the electrophilic carbonyl carbon atom to create a tetrahedral intermediate. This tetrahedral intermediate then undergoes an elimination to yield the products. In this two-stage mechanism bond formation occurs before bond cleavage, and the carbonyl carbon atom undergoes a hybridization change from sp2 to sp3 and back again. The facility with which nucleophilic reagents add to a carbonyl group was noted earlier for aldehydes and ketones.
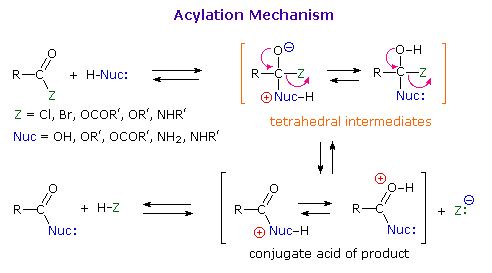
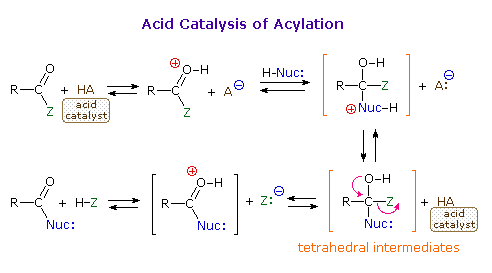
Acid and base-catalyzed variations of this mechanism are shown above. Also, a specific example of acyl chloride formation from the reaction of a carboxylic acid with thionyl chloride will be shown. The number of individual steps in these mechanisms vary, but the essential characteristic of the overall transformation is that of addition followed by elimination. Acid catalysts act to increase the electrophilicity of the acyl reactant; whereas, base catalysts act on the nucleophilic reactant to increase its reactivity. In principle all steps are reversible, but in practice many reactions of this kind are irreversible unless changes in the reactants and conditions are made. The acid-catalyzed formation of esters from carboxylic acids and alcohols, described earlier, is a good example of a reversible acylation reaction, the products being determined by the addition or removal of water from the system. The reaction of an acyl chloride with an alcohol also gives an ester, but this conversion cannot be reversed by adding HCl to the reaction mixture.
|
Mechanisms of Ester Cleavage Esters are one of the most common carboxylic derivatives. Cleavage of the alkyl moiety in an ester may be effected in several different ways, the most common being the acyl transfer mechanism described above; however, other mechanisms have been observed. |
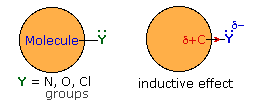 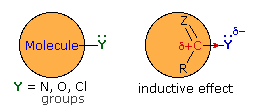 |
Thus far we have not explained the marked variation, noted above, in the reactivity of different carboxylic acid derivatives. The distinguishing carbonyl substituents in these compounds are: chloro (acyl chlorides), acyloxy (anhydrides), alkoxy (esters) and amino (amides). All of these substituents have bonds originating from atoms of relatively high electronegativity (Cl, O & N). They are therefore inductively electron withdrawing when bonded to carbon, as shown in the diagram on the right. The consequences of such inductive electron withdrawal on the acidity of carboxylic acids was previously noted.
When these substituents are attached to an sp2 carbon that is part of a π-electron system, a similar inductive effect occurs, but n-π conjugation (p-π conjugation) moves electron density in the opposite direction. By clicking the "Toggle Effect" button the electron shift in both effects will be displayed sequentially. This competition between inductive electron withdrawal and conjugative electron donation was discussed earlier in the context of substituent effects on electrophilic aromatic substitution. Here, it was noted that amino groups were strongly electron donating (resonance effect >> inductive effect), alkoxy groups were slightly less activating, acyloxy groups still less activating (resonance effect > inductive effect) and chlorine was deactivating (inductive effect > resonance effect). In the illustration on the right, R and Z represent the remainder of a benzene ring.
This analysis also predicts the influence these substituent groups have on the reactivity of carboxylic acid derivatives toward nucleophiles (Z = O in the illustration). Inductive electron withdrawal by Y increases the electrophilic character of the carbonyl carbon, and increases its reactivity toward nucleophiles. Thus, acyl chlorides (Y = Cl) are the most reactive of the derivatives. Resonance electron donation by Y decreases the electrophilic character of the carbonyl carbon. The strongest resonance effect occurs in amides, which exhibit substantial carbon-nitrogen double bond character and are the least reactive of the derivatives. An interesting exception to the low reactivity of amides is found in beta-lactams such as penicillin G. The angle strain introduced by the four-membered ring reduces the importance of resonance, the non-bonding electron pair remaining localized on the pyramidally shaped nitrogen. Finally, anhydrides and esters have intermediate reactivities, with anhydrides being more reactive than esters.
|
Carbonyl Reactivity and IR Stretching Frequency |
|
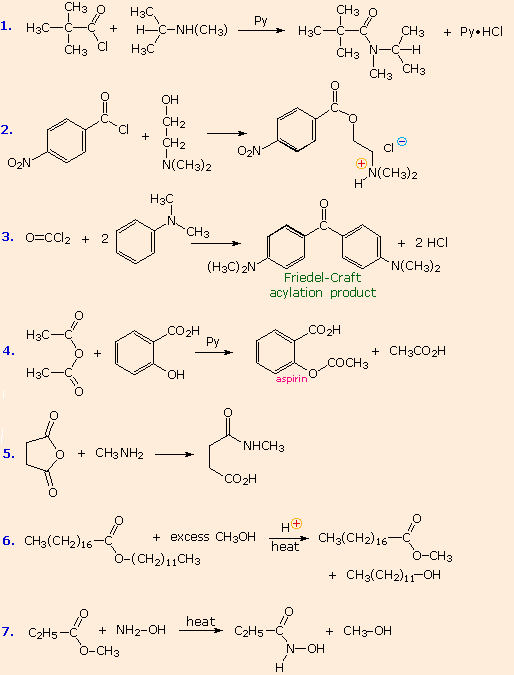
From the previous discussions you should be able to predict the favored product from each of the following reactions. |
||
|
 |
|
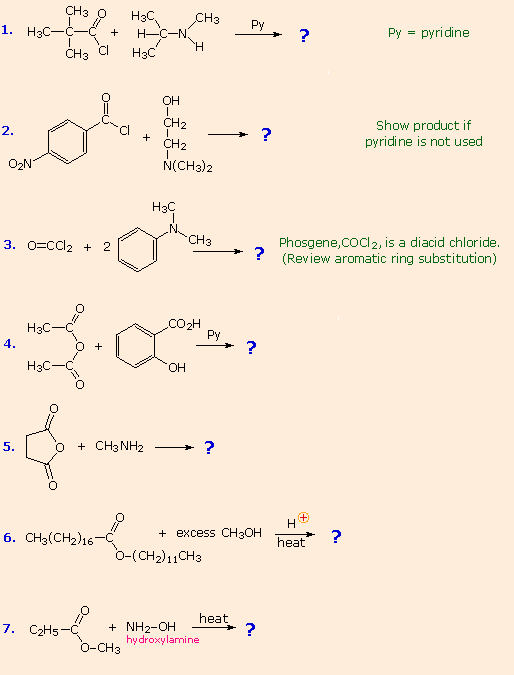
The first three examples concern reactions of acyl chlorides, the most reactive acylating reagents discussed here. Although amines are among the most reactive nucleophiles, only 1º and 2º-amines give stable amide products. Reaction of 3º-amines with strong acylating reagents may generate acylammonium species reversibly (see below), but these are as reactive as acyl chlorides and will have only a very short existence. This explains why reactions #2 & 3 do not give amide products.
| RCOCl | + | R'3N |  |
RCONR'3(+) Cl(–) (an acylammonium salt) |
Reactions #4 & 5 display the acylating capability of anhydrides. Bear in mind that anhydrides may also be used as reagents in Friedel-Crafts acylation reactions. Esters are less reactive acylating reagents than anhydrides, and the ester exchange reaction (#6) requires a strong acid or base catalyst. The last example demonstrates that nitrogen is generally more nucleophilic than oxygen. Indeed, it is often possible to carry out reactions of amines with acyl chlorides and anhydrides in aqueous sodium hydroxide solution! Not only is the amine more nucleophilic than water, but the acylating reagent is generally not soluble in or miscible with water, reducing the rate of its hydrolysis.
No acylation reactions of amides were shown in these problems. The most important such reaction is hydrolysis, and this normally requires heat and strong acid or base catalysts. One example, illustrating both types of catalysis, is shown here. Mechanisms for catalyzed reactions of this kind were presented earlier.
| R–CO2(–) + CH3NH2 | OH(–) & heat |
|
H(+) & heat |
R–CO2H + CH3NH3(+) |
|
Other Acylation Reagents and Techniques |
Nitriles
Although they do not have a carbonyl group, nitriles are often treated as derivatives of carboxylic acids. Hydrolysis of nitriles to carboxylic acids was described earlier, and requires reaction conditions (catalysts and heat) similar to those needed to hydrolyze amides. This is not surprising, since addition of water to the carbon-nitrogen triple bond gives an imino intermediate which tautomerizes to an amide.
R–C≡N + H2O |
acid or base |
R–C(OH)=NH |
 |
R–CO–NH2 |
Contributors
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry