
7.6: Carbon acids
- Page ID
- 21360

So far, we have limited our discussion of acidity and basicity to heteroatom acids, where the acidic proton is bound to an oxygen, nitrogen, sulfur, or halogen. However, carbon acids - in which the proton to be donated is bonded to a carbon atom - play an integral role in biochemistry.
A: The acidity of alpha protons
A hydrogen on an alkane is not at all acidic – its pKa is somewhere on the order of 50, about as non-acidic as it gets in the organic chemistry world. The reason for this is that if the hydrogen were to be abstracted, the electrons from the broken bond would be localized on a single carbon atom.
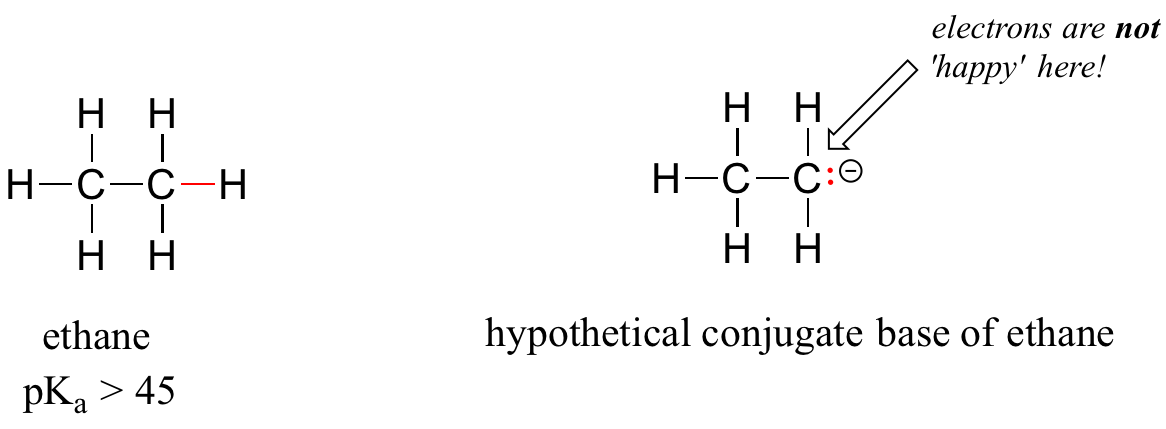
Because carbon is not electronegative and is terrible at holding a negative charge, such carbanion species are extremely unstable.
How, then, can a proton bonded to a carbon be acidic? Remember that an acid becomes stronger if the conjugate base is stabilized, an in particular if the negative charge on the conjugate base can be delocalized to an electronegative atom such as an oxygen. This is possible when a carbon is located adjacent to a carbonyl group. Consider, for example, the conjugate base of acetone.
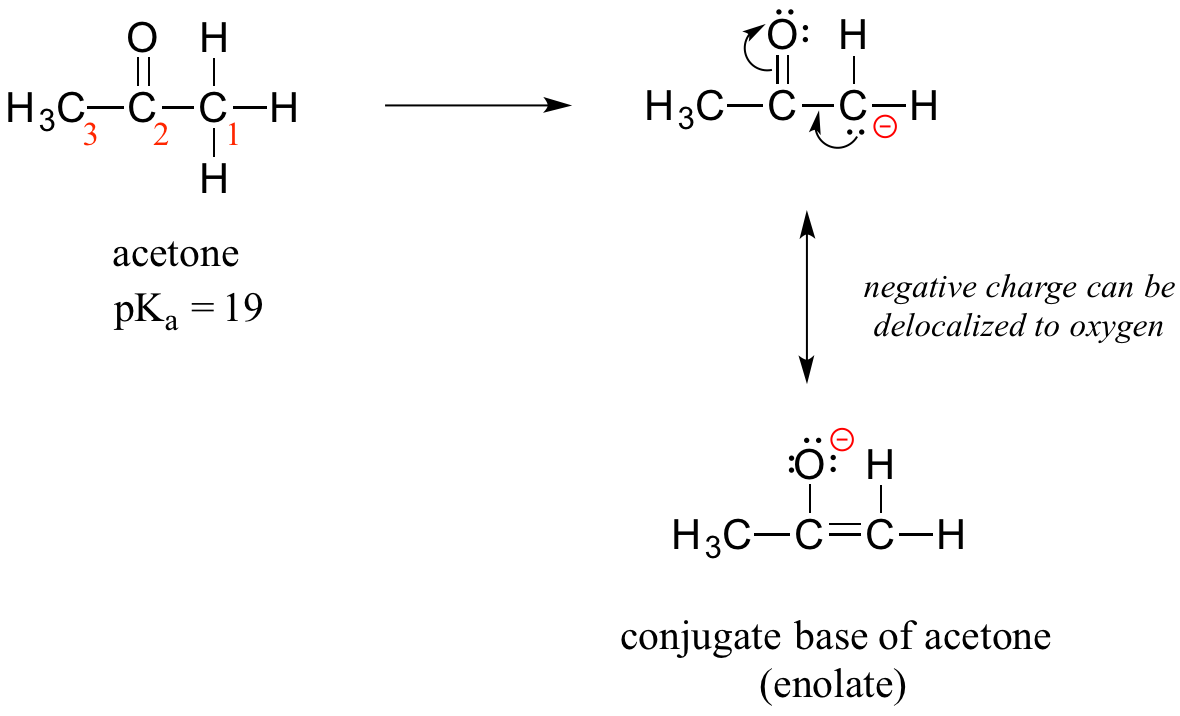
One resonance contributor puts the negative charge on the carbon #1. Due to the presence of the adjacent carbonyl group, however, a second resonance contributor can be drawn in which the negative charge is located on the carbonyl oxygen, where it is much more stable. This type of stabilized carbanion species is specifically referred to as an enolate. Acetone is in fact weakly acidic, with a pKa of about 19. The importance of the position of the carbonyl group is evident when we consider 2-butanone: here, the protons on carbons #1 and #3 are somewhat acidic (in the neighborhood of pKa = 20), but the protons on carbon #4 are not acidic at all, because carbon #4 is not adjacent to the carbonyl.
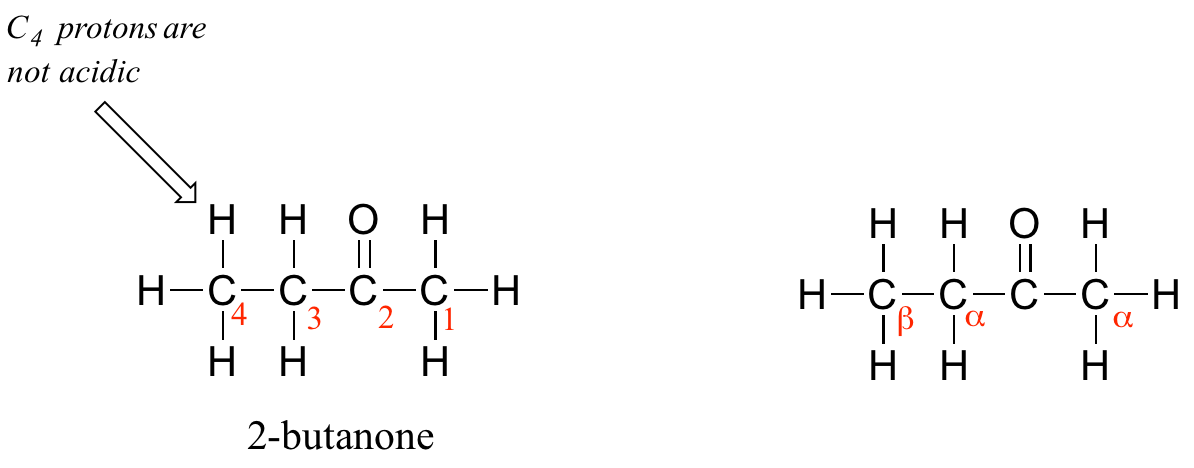
A carbon that is located next to a carbonyl group is referred to as an alpha (α) carbon, and any proton bound to it is an alpha-proton. In 2-butanone, carbons #1 and #3 are alpha carbons, and their five protons are alpha protons. Carbon #4 is a beta (β) carbon, as it is two positions removed from the carbonyl carbon
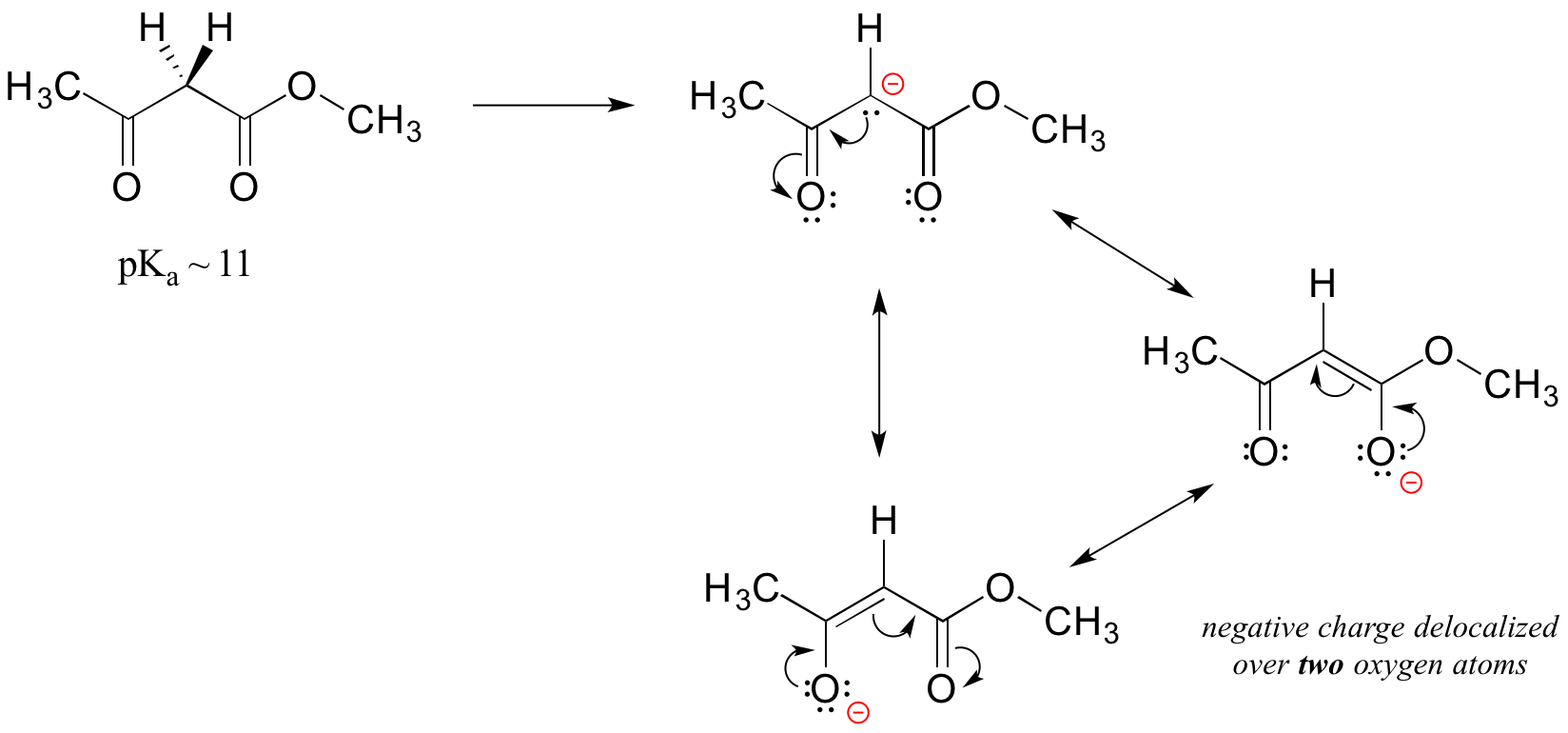
An active methylene is a carbon in the alpha position relative to two carbonyl groups rather than just one. Protons on active methylene carbons are more acidic than other alpha protons, because the charge on the conjugate base can be localized to two different oxygen atoms, not just one. This keto-ester compound, for example, has a pKa of approximately 11, close to that of phenol.
As we alluded to above, the acidity of alpha protons is an extremely important concept in biological organic chemistry. Look through a biochemistry textbook, and you will see reaction after reaction in which the first mechanistic step is the abstraction of an alpha proton to form an enolate intermediate. Two chapters in this book (chapters 12 and 13) are devoted to such reactions, and the initial proton-extraction step of three example reactions are previewed below. Reaction A is from fatty acid oxidation, while reactions B and C are both part of carbohydrate metabolism.
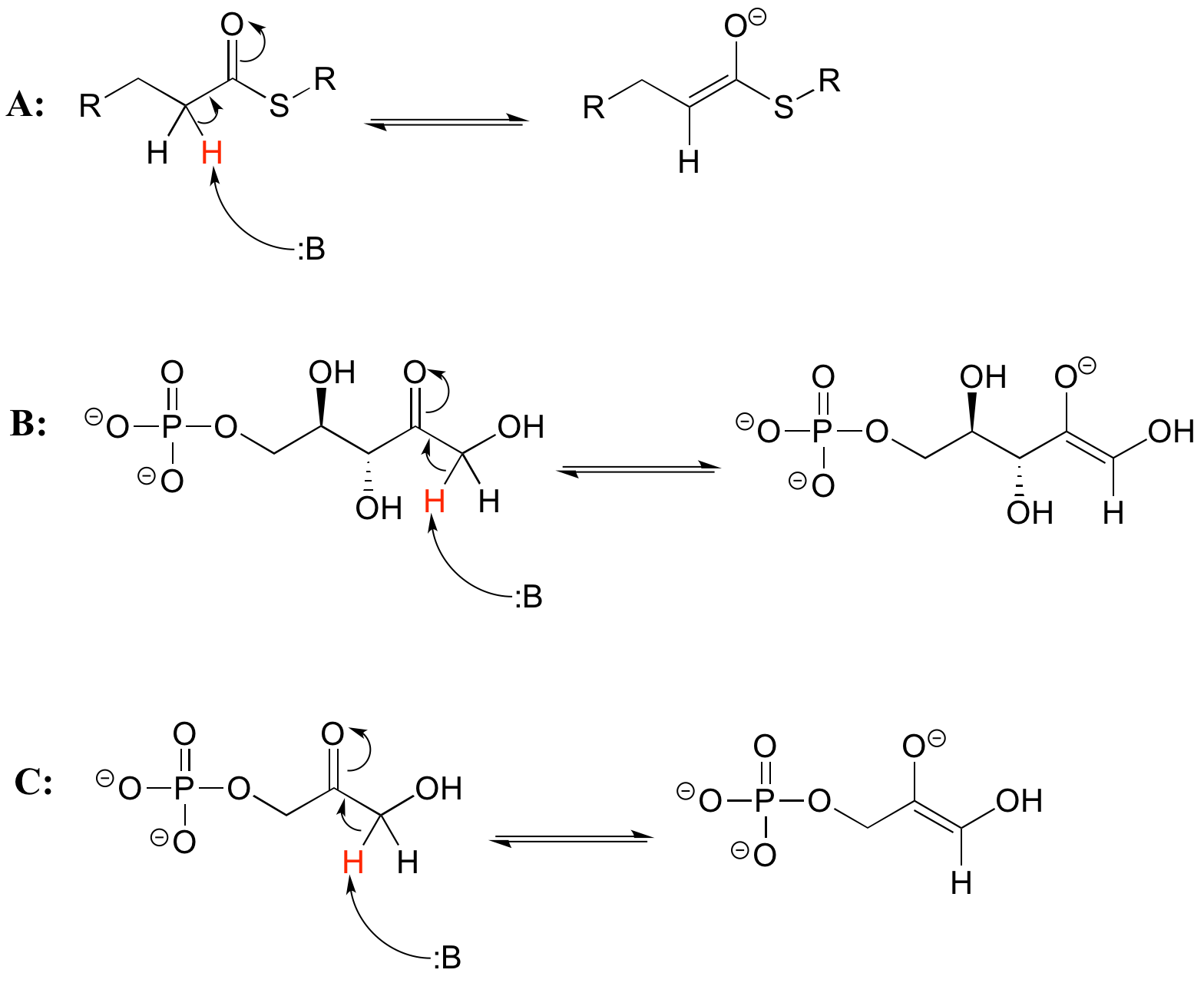
Exercise 7.6.1
For each molecule shown below,
a) Show the location of all alpha protons.
b) Draw the structure(s) of all possible enolate conjugate bases.

B: Keto-enol tautomers
An enolate ion can, of course, be reprotonated at the alpha carbon to return the molecule to the ketone or aldehyde form. An alternate possibility is that the oxygen atom, rather than the alpha carbon, could be protonated. The species that results from this step is referred to as an enol (this term reflects the fact that an enol contains structural elements of both an aklene and an alcohol).
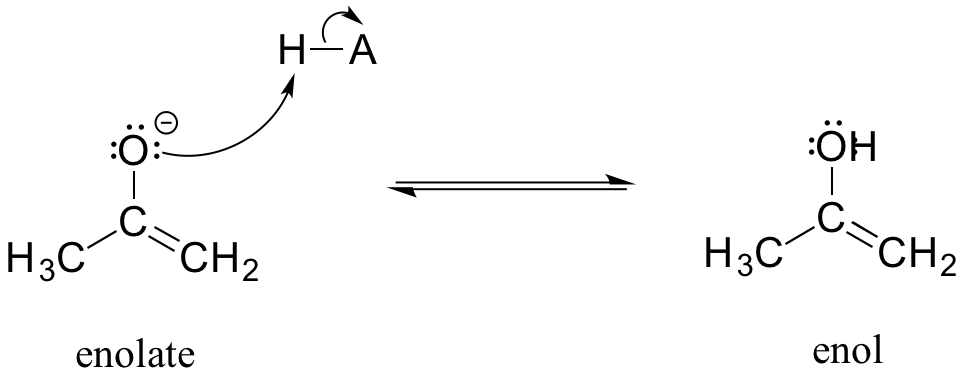
In fact, most ketones and aldehydes exist in rapid equilibrium with their enol form. A ketone/aldehyde and its corresponding enol are tautomers: a pair of constitutional isomers which can be rapidly and reversibly interconverted, and which vary in terms of the site of protonation and location of a double bond. As we will see going forward, tautomerization - the interconversion of two tautomers - is a ubiquitous step in biological organic chemistry. Often, when discussing tautomerization, the ketone (or aldehyde) isomer is referred as the keto form.
As a general rule, the keto form is lower in energy than the corresponding enol form, and thus the keto form predominates at equilibrium. Acetone, for example, is present at >99% keto form at equilibrium, and the enol form at less than 1%.
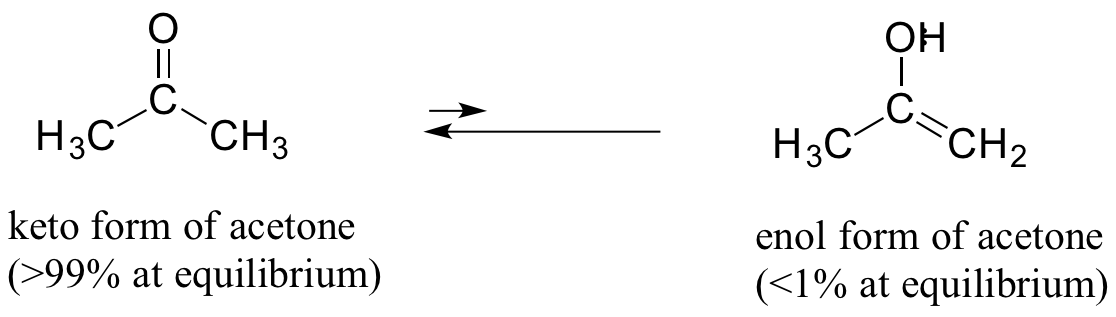
The 'driving force' for the enol to keto conversion can be understood in terms of the energies of the three bonds involved in the process: the sum of the three bond energies is about 48 kJ/mol greater in the keto form than in the enol form.

Exercise 7.6.2
Draw all of the possible enol forms of the following aldehydes/ketones. There may be more than one possible enol form (consider stereochemistry as well as regiochemistry!)
a) 3-pentanone
b) acetaldehyde (IUPAC name ethaldehyde)
c) cyclohexanone
d) 2-pentanone
Exercise 7.6.3
Draw three examples of aldehyde or ketone compounds for which there is no possible enol form.
Exercise 7.6.4
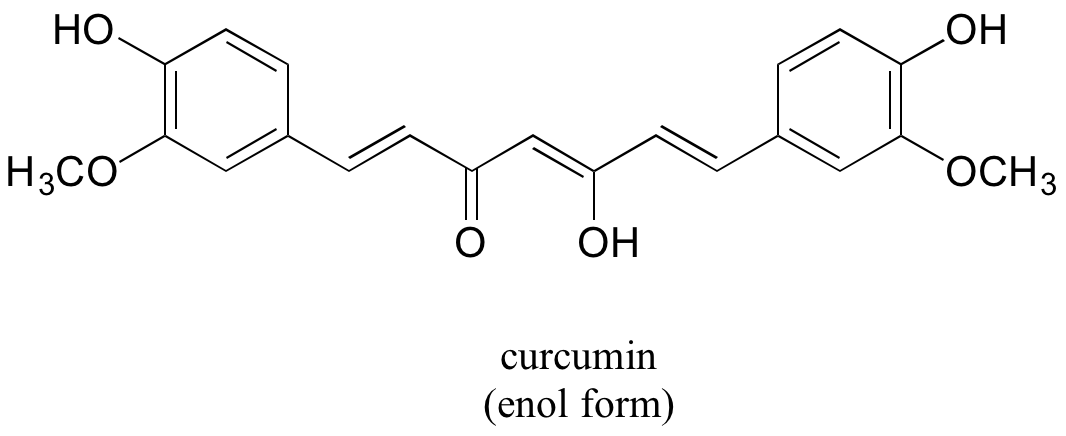
In some special cases, the enol form of a compound is more stable than the keto form and thus predominates at equilibrium. Curcumin is the compound mainly responsible for the characteristic yellowish color of tumeric, a ubiquitous spice in indian cuisine. The extended system of p bonds present in the enol form causes it to be lower in energy than the keto form. Draw the keto form of curcumin, and explain how the conjugated pi system is disrupted in this form.
Exercise 7.6.5
The phenol functional group can also be thought of a kind of enol.
a) Draw the 'keto' form of phenol.
b) The 'keto' form of phenol is highly disfavored compared to the 'enol' form - why?
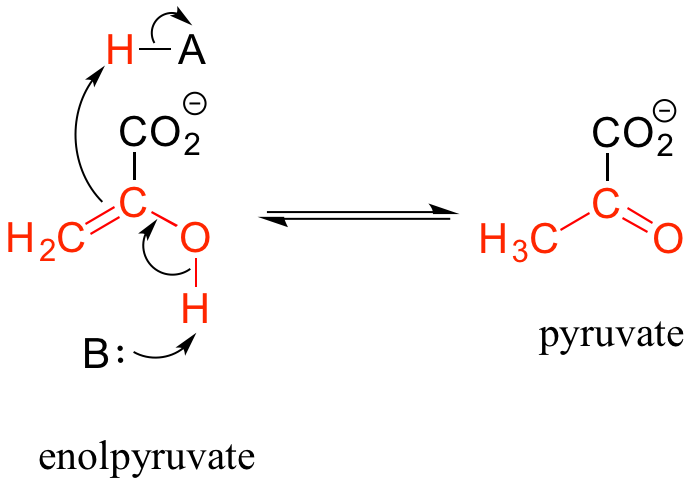
Keto-enol tautomerization steps can be found in many biochemical reactions. For example, there is an enol to keto tautomerization step in the glycolysis reaction catalyzed by pyruvate kinase (EC 2.7.1.40). Shown below is just the tautomerization part of this reaction; we will see the complete reaction in chapter 9.
C: Imine-enamine tautomers
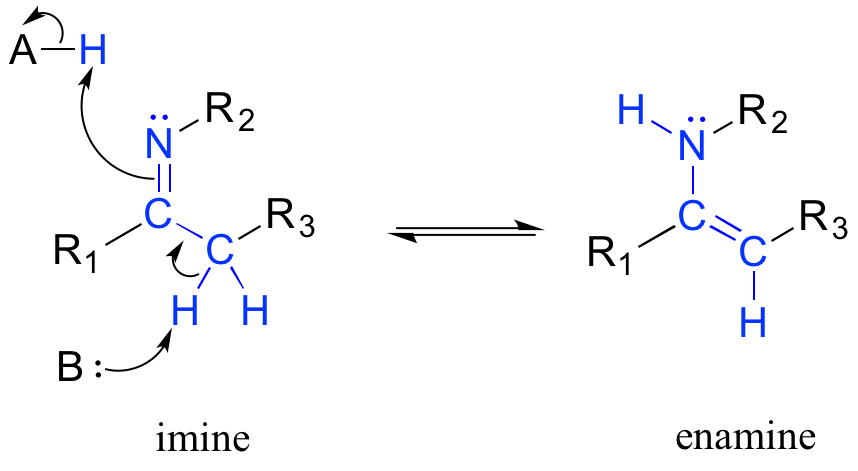
Another common tautomeric relationship in biological organic chemistry is the equilibrium between imines (also known as Schiff bases) and enamines, which are essentially the nitrogen equivalents of enols.
Mechanism for imine - enamine tautomerization
The degradation of serine, for example, involves an enamine to imine tautomerization step: (EC 4.2.1.13)
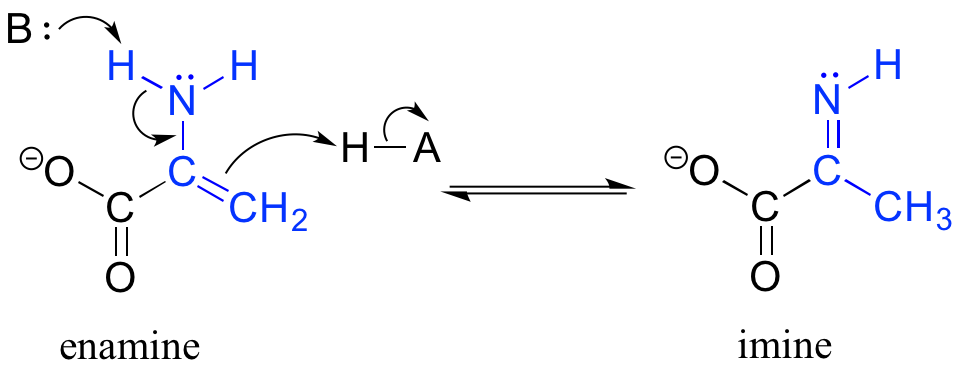
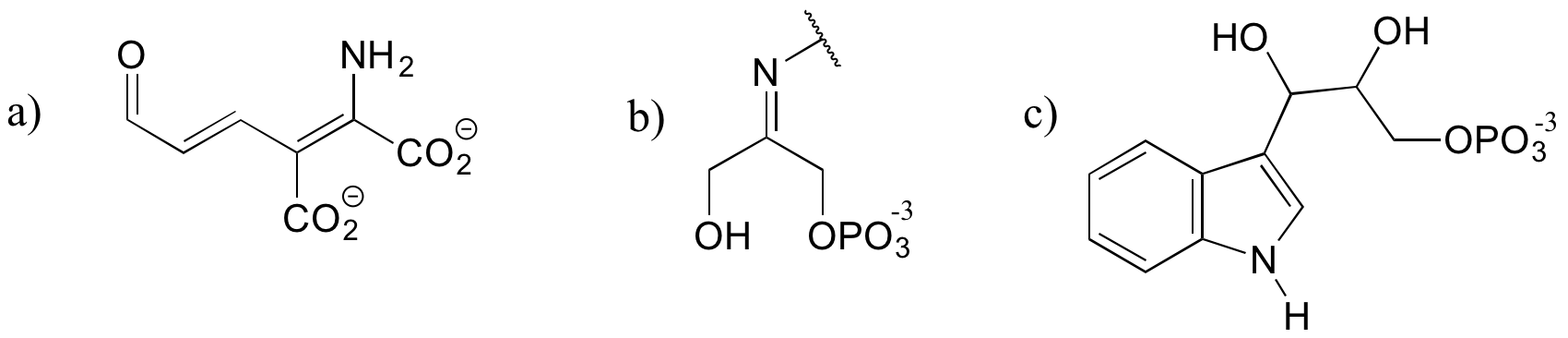
Exercise 7.6.6
The structures below all contain either an imine or an enamine group. For each, draw the structure of an alternate tautomer.
D: Acidity of terminal alkynes
Terminal alkynes are another kind of carbon acid which are relevant more to laboratory organic chemistry than to biological chemistry

Terminal alkynes are more acidic than alkenes or alkanes for the same reason that protonated imines are more acidic than protonated amines: the alkyne carbon is sp-hybridized, meaning that it has 50% s-orbital character and is therefore more electronegative. With a pKa of approximately 26, alkynes are only weakly acidic, but nonetheless can be fully deprotonated through the use of a strong base such as sodium amide (NaNH2).
Exercise 7.6.7
Hydrogen cyanide, HCN, is another example of a relatively strong carbon acid, with a pKa of 9.2. Suggest a rationale for the acidity of this proton.