
14.5: Thiamine diphosphate-dependent reactions
- Page ID
- 989

Thiamine diphosphate (TPP) is another very important coenzyme which, like PLP, acts as an electron sink to stabilize key carbanion intermediates. The important part of the TPP molecule from a catalytic standpoint is its thiazole ring.

14.5A: The benzoin condensation reaction
In a very simple experiment that can performed in an undergraduate organic chemistry lab, benzaldehyde (a liquid compound at room temperature that is used as an artificial cherry flavoring) self-condenses to form benzoin (a crystalline solid) when stirred in ethanol with a catalytic amount of sodium hydroxide and thiamine.
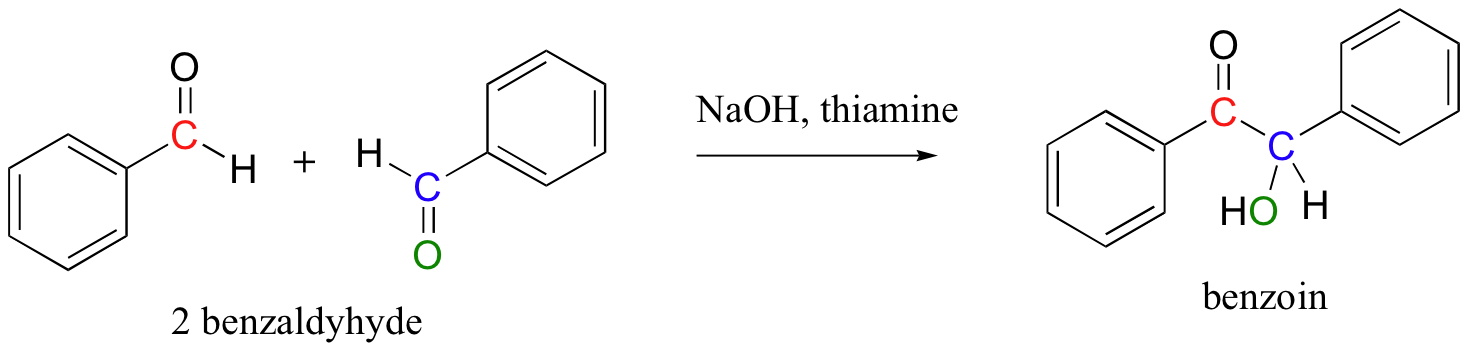
The laboratory synthesis of benzoin is interesting because it mimics the mechanism of TPP-dependant enzymatic reactions in biological systems. This reaction is not catalyzed by an enzyme – rather, the thiamine molecule acts on its own, playing a similar catalytic role to that played by its diphosphate ester cousin (TPP) in enzymatic reactions.
Look carefully at the connectivity of the starting compounds and product in the benzoin condensation. Essentially what has happened is that the carbonyl carbon of one benzaldehyde molecule has somehow been turned into a nucleophile, and has attacked a second benzaldehyde molecule in a nucleophilic carbonyl addition reaction.

This probably seems quite strange - we know that carbonyl carbons are good electrophiles, but how can they be nucleophilic? For the aldehyde carbon to be a nucleophile, it would have to be deprotonated, and become a carbonyl anion. But aldehyde protons are not at all acidic!
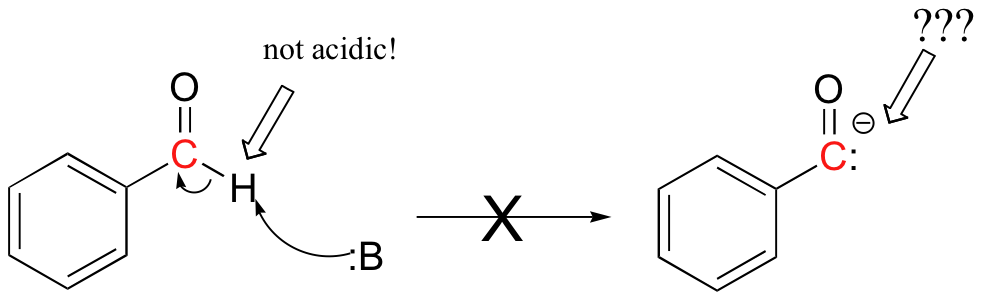
The thiamine catalyst is the key: it allows the formation of what is essentially the equivalent of a nucleophilic benzaldehyde carbanion. Let's follow the benzoin condensation reaction mechanism through step-by-step, and see how thiamine accomplishes this task.
The important part of the thiamine molecule is the thiazole ring (look again at the structure of thiamine diphosphate on the previous page), thus we will draw thiamine (and later, thiamine diphosphate) using R groups to depict the unreactive parts of the molecule. The first step of the benzoin condensation is deprotonation of thiamine by hydroxide.
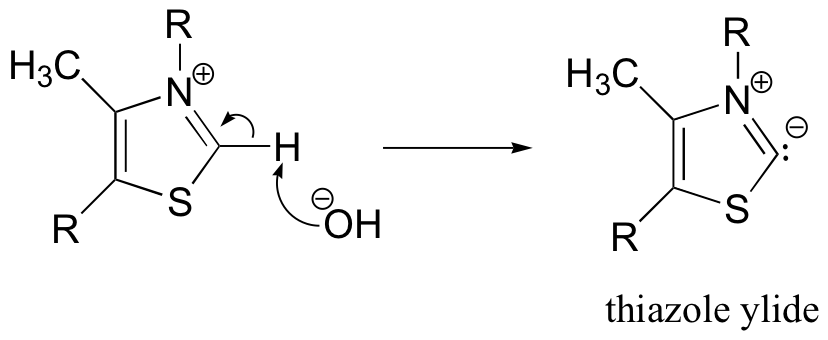
It may surprise you to learn that this proton is acidic. The reason for its acidity lies partly in the ability of the neighboring sulfur atom to accept, in its open d orbitals, some of the excess electron density of the conjugate base. Another reason is that the positive charge on the nitrogen helps to stabilize the negative charge on the conjugate base. The deprotonated thiazole is called an ylide, which is a species with adjacent positively and negatively charged atoms.
The negatively charged carbon on the thiazole ylide next attacks the carbonyl of the first benzaldehyde molecule in a nucleophilic carbonyl addition (from here on out, thiamine will be colored green in order to help you to focus on the chemistry going on with the substrate).
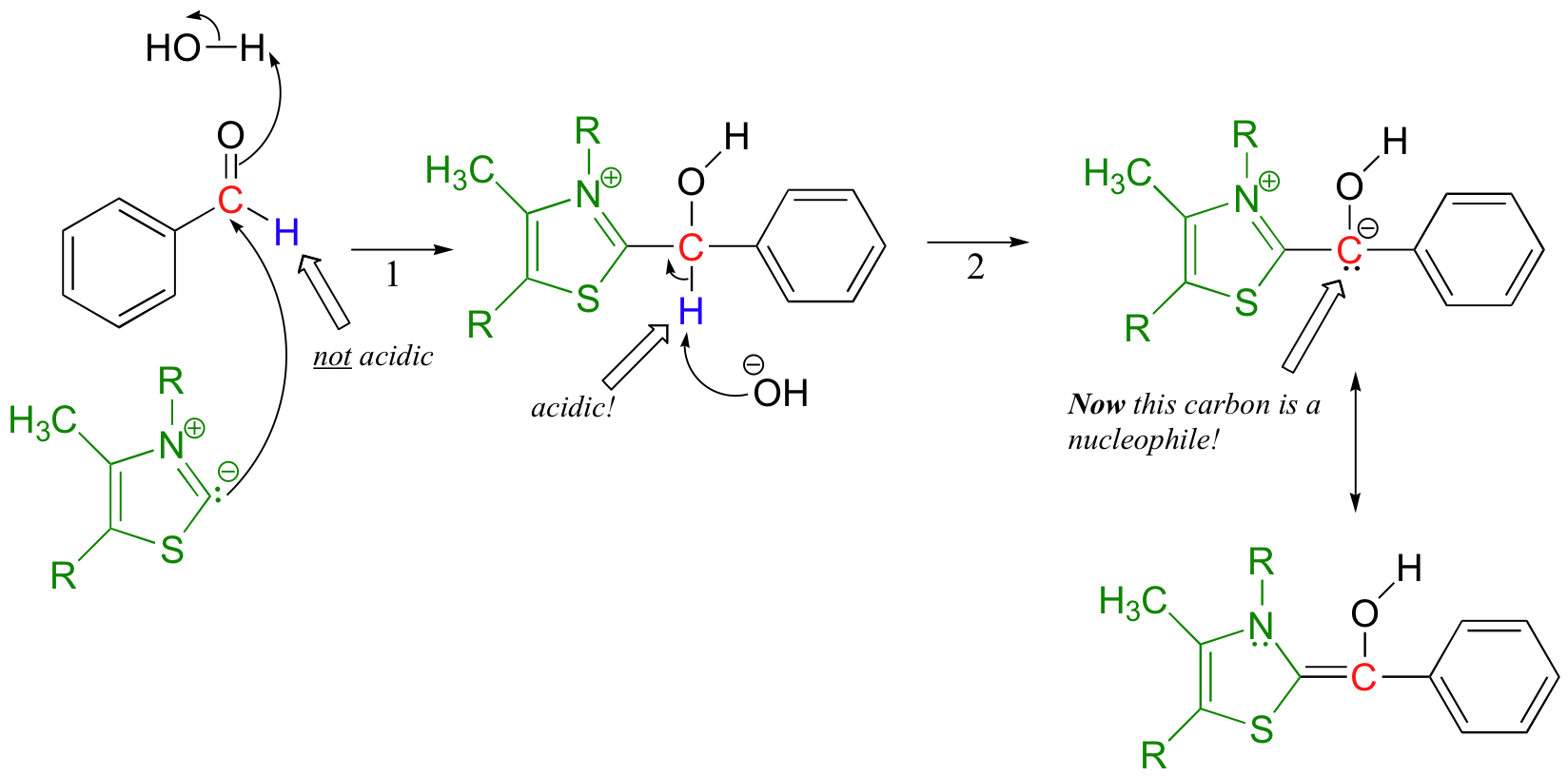
In the resulting molecule, what used to be the aldehyde hydrogen is now acidic - this is so because, when the proton is abstracted by hydroxide, the negative charge on the conjugate base is stabilized by resonance with the positively-charged thiazole ring of thiamine. This is the function of thiamine: it acts as an electron sink, accepting electron density so as to allow for the formation of what amounts to a carbonyl anion.
Now the first benzaldehyde molecule, assisted by thiamine, can finally act as a nucleophile, attacking the carbonyl of a second benzaldehyde (step 3).
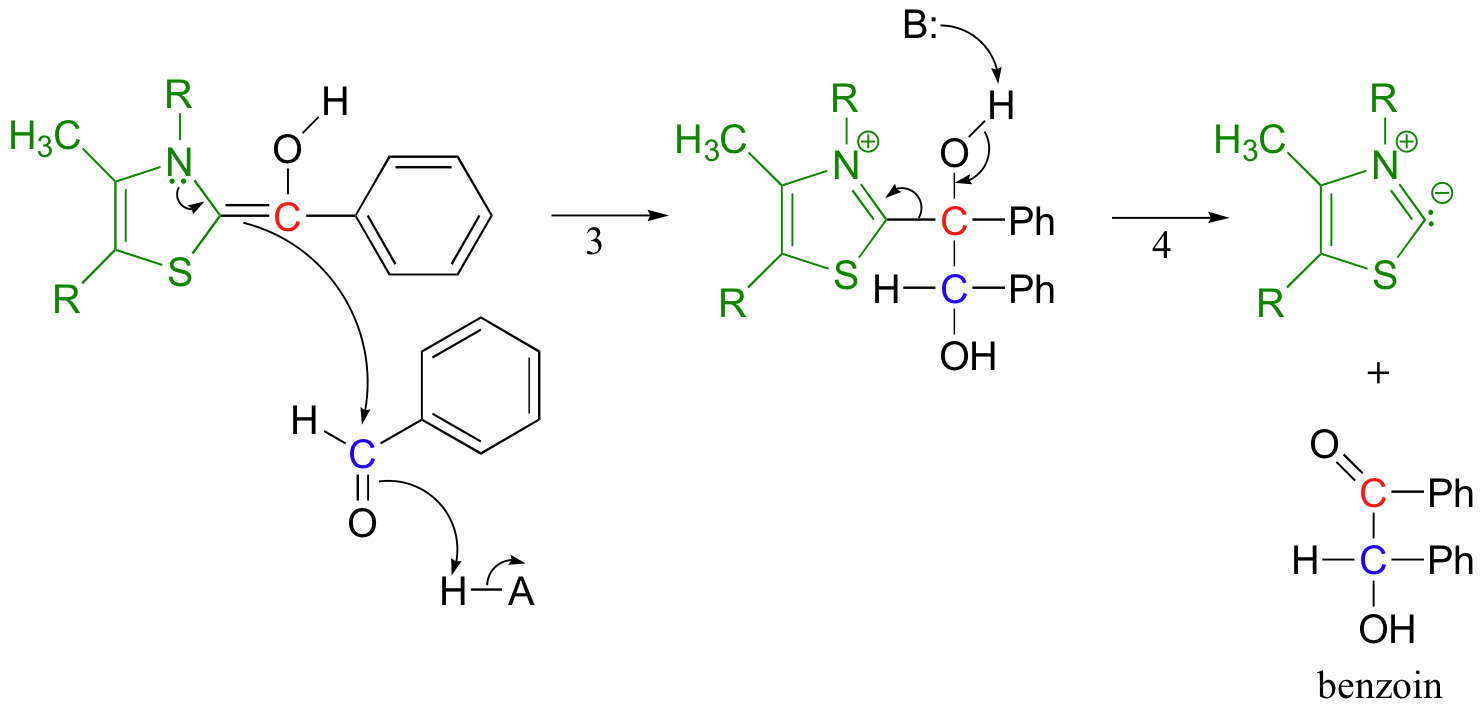
Once this is accomplished, the thiamine ylide can be kicked off as the original carbonyl on the first benzaldehyde re-forms (step 4). The thiamine ylide is now free to catalyze another reaction.
14.5B: The transketolase reaction
Now let's look at a biochemical reaction carried out by an enzyme called transketolase, with the assistance of thiamine diphosphate. Transketolase is one of a series of enzymes (along with ribulose-5-phosphate-3-epimerase, which we considered in section 13.2B) in the 'Calvin cycle' of carbon fixation in plants. Animals and bacteria also use transketolase in sugar metabolism. In the transketolase reaction, a 2-carbon unit (in the smaller, red box) is transferred between two sugar molecules:

First, let's consider how this reaction might hypothetically proceed without the assistance of the TPP cofactor. The breaking off of the 2-carbon unit from fructose-6-phosphate might be depicted as a kind of retro-aldol event:
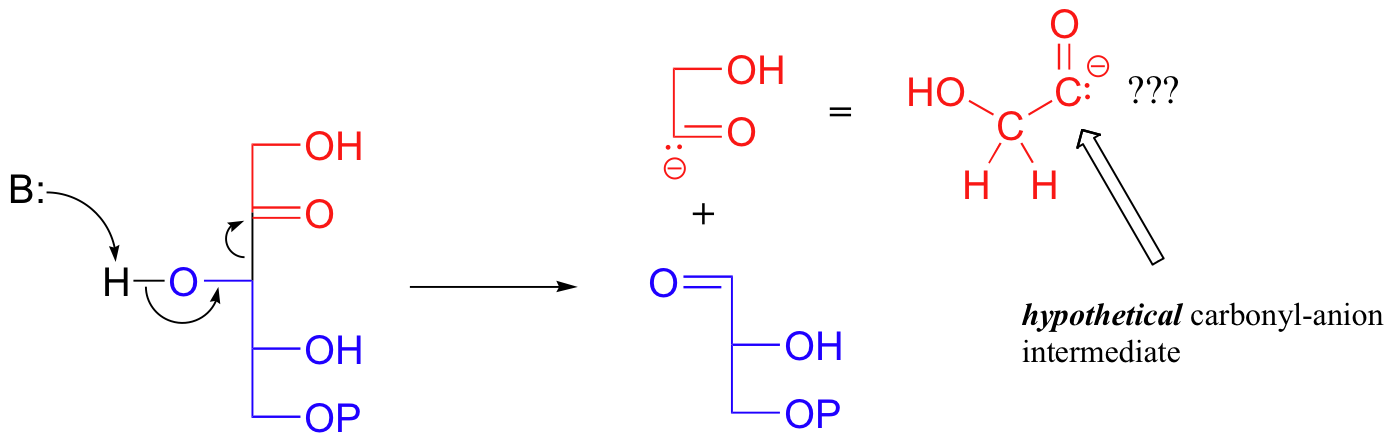
This would lead to glyceraldehyde-3-phosphate, the first product, plus an intermediate in the form of a carbonyl anion.
Continuing to think hypothetically, this strange 2-carbon ionic intermediate could attack the glyceraldehyde-3-phosphate carbonyl, resulting in sedoheptulose-7-phosphate, the second product.
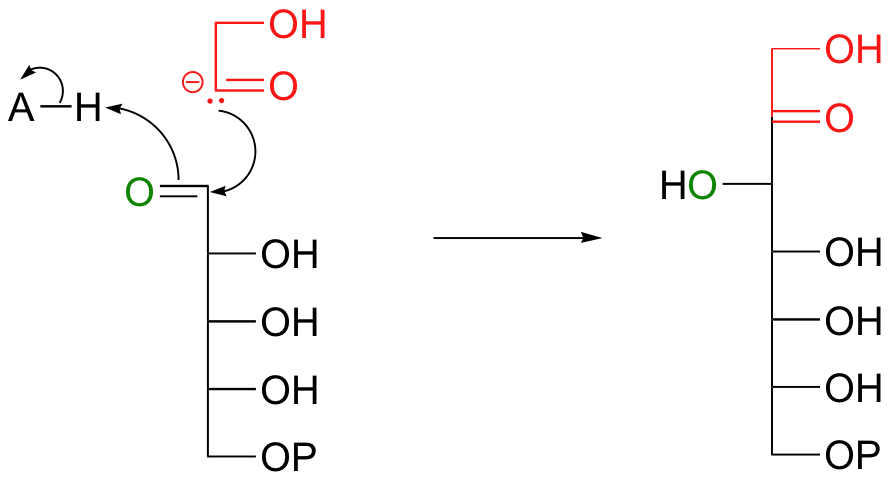
Everything in this hypothetical scenario is fine, chemically speaking, with one major exception: the carbonyl anion intermediate. It would be an extremely high energy, unlikely species. But we have seen something like this before, haven't we, in the non-enzymatic benzoin condensation reaction above! This is exactly the kind of carbanion that thiamine (this time in its diphosphate form, TPP) makes possible. The carbonyl anion is generated in different ways in the two reactions: in the benzoin condensation it is the result of the deprotonation of benzaldehyde, while in the transketolase it results from a retro-aldol-like cleavage. But that doesn't really matter - what does matter is that in each case, a thiazole ring is present to modify the starting carbonyl group and act as an electron sink, allowing it to take on a negative charge. Here, then, is the real (as opposed to hypothetical) transketolase reaction, with the role of TPP revealed.
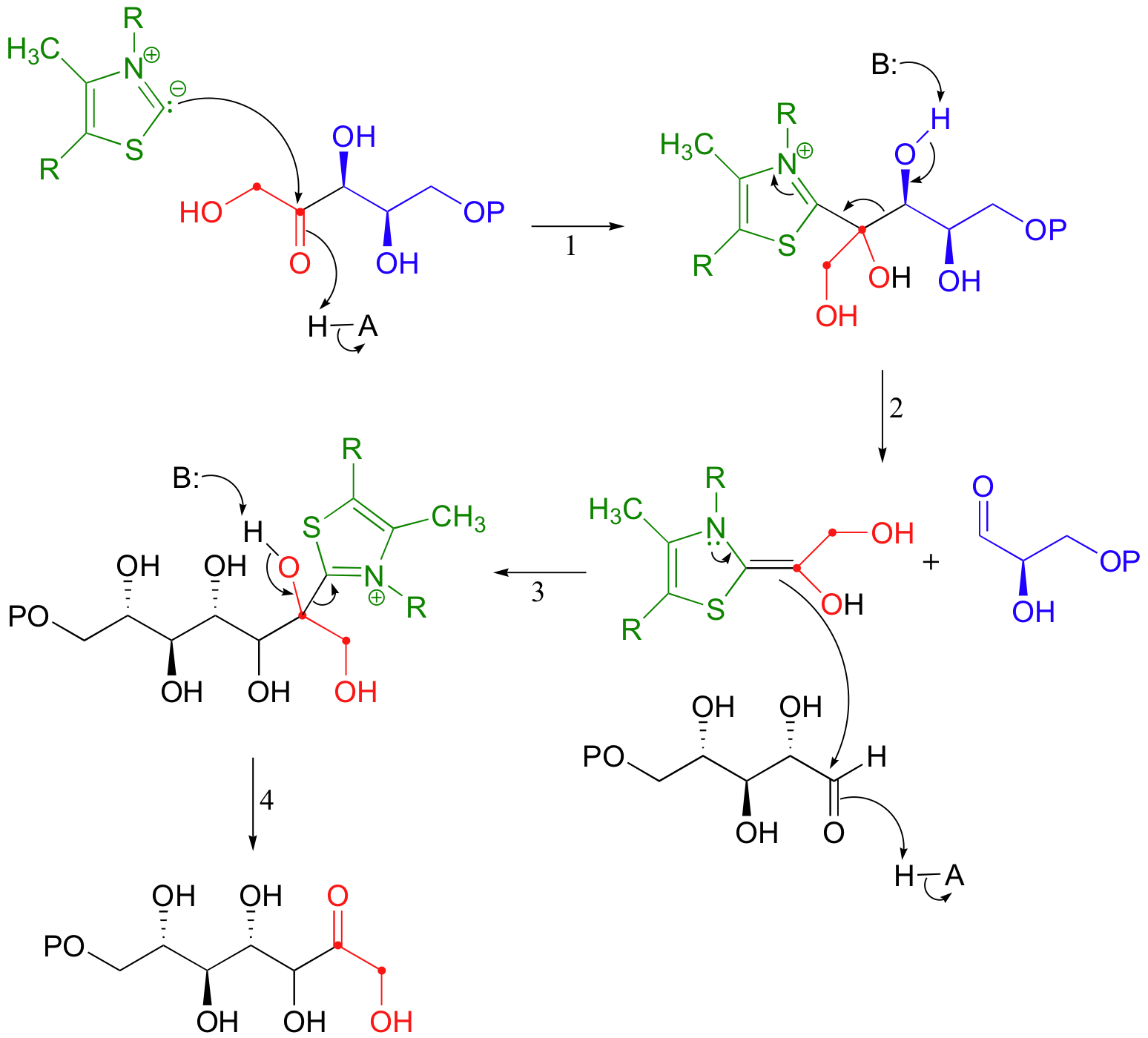
Make sure that you can follow the electron movement throughout the mechanism, that you can see how TPP acts as an electron sink cofactor, and that you clearly recognize the mechanistic parallels to the benzoin condensation.
14.5C: Pyruvate decarboxylase
The thiamine diphosphate coenzyme also assists in the decarboxylation of an acyl group, such as in this reaction catalyzed by pyruvate decarboxylase (this is a key reaction in the fermentation of glucose to ethanol by yeast):

In this example, the TPP-stabilized carbonyl carbanion simply acts as a base rather than as a nucleophile, abstracting a proton from an enzymatic acid to form acetaldehyde.
Example 14.7
Propose a mechanism for the reaction catalyzed by pyruvate decarboxylase.
14.5D: Synthetic parallel - carbonyl nucleophiles via dithiane anions
Nature uses thiamine to generate the equivalent of a nucleophilic carbonyl anion, but with the specific exception of the benzoin condensation, a chemist working in an organic synthesis laboratory before the mid-1970's had no equivalent procedure. In 1975, E. J. Corey and Dieter Seebach reported that they had developed a method to accomplish reactions such as the following, in which aldehyde carbons act as nucleophiles in SN2 and carbonyl addition reactions (J. Org. Chem. 1975, 40, 231).
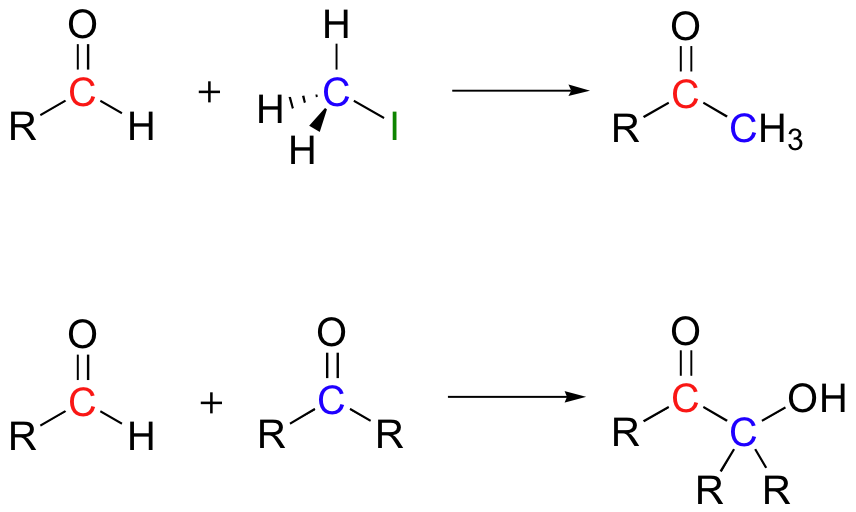
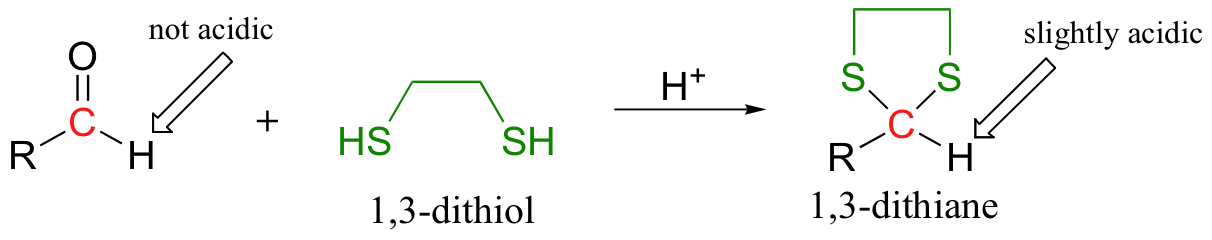
In this technique, the aldehyde is first converted to a cyclic thioketal using 1,3-propanedithiol and acid catalyst - this is the same as the method used to 'protect' aldehydes as cyclic acetals, discussed in section 11.4B, except that a dithiol is used instead of a diol.
The original aldehyde proton is now somewhat acidic, because the empty d orbitals on the two adjacent sulfur atoms are able to delocalize excess electron density of the conjugate base. A strong base, such as an organolithium compound (section 13.6B) will deprotonate the cyclic thioacetal, which can then act as a nucleophile, attacking an alkyl halide or a carbonyl electrophile (the latter case is illustrated below):
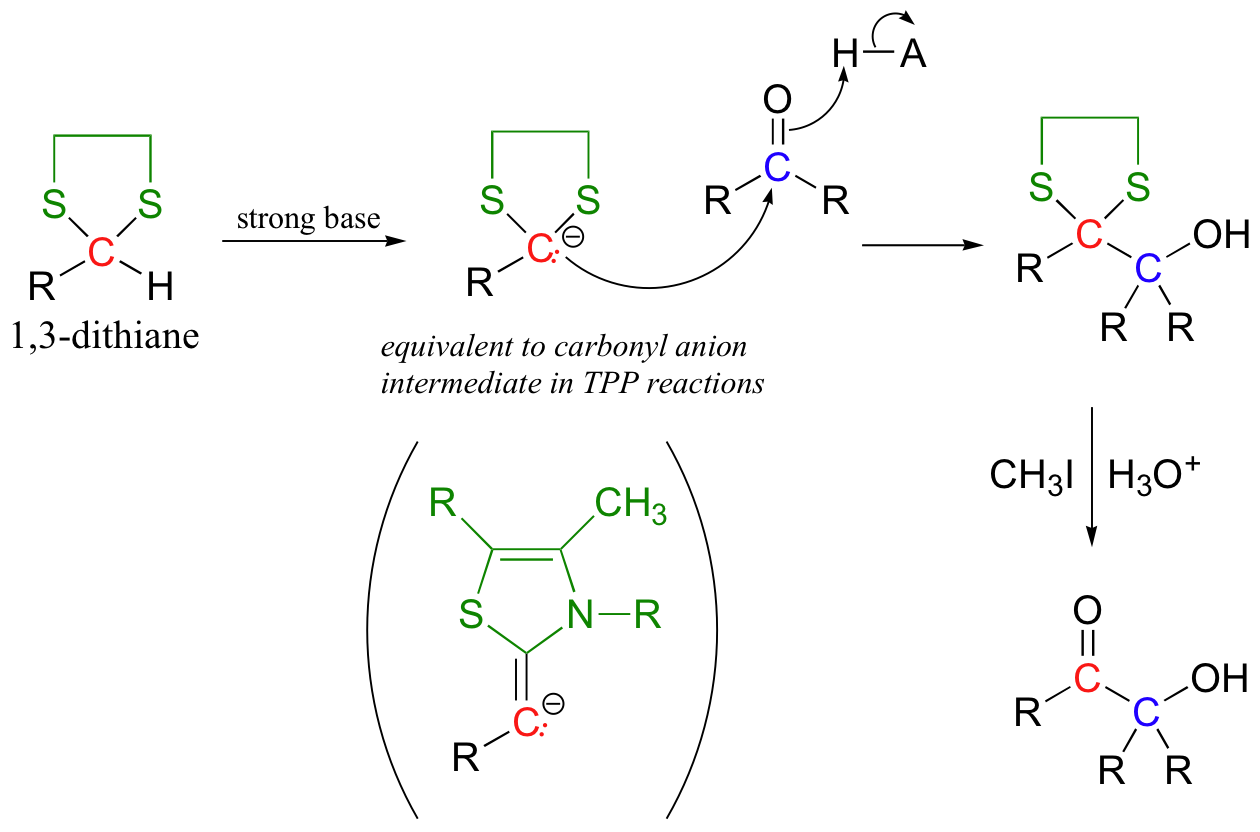
The thioacetal can then be hydrolyzed back to an aldehyde group, a process that is facilitated by the use of methyl iodide.
Example 14.8