
II: Transition-Metal-Generated Radicals
- Page ID
- 23811
Reactions of radicals generated from transition-metal complexes can be divided into two types based on the direction of electron flow. In some of these reactions the transition metal accepts an electron during radical formation (oxidative electron transfer) and in others it donates an electron during this process (reductive electron transfer). The compounds that most often participate in oxidative electron transfer are manganese(III) acetate [Mn(OAc)3] and ammonium cerium(IV) nitrate [(NH4)2Ce(NO3)6], while those frequently involved in reductive electron transfer are bis(cyclopentadienyl)titanium(III) chloride (Cp2TiCl), and samarium(II) iodide (SmI2). Carbohydrates that are bonded to a cobalt-containing complex by a C–Co bond form radicals by oxidative electron transfer and then frequently reform a C–Co bond by reductive electron transfer.
Coenzyme B12 (5, Figure 1) is one of a group of biologically active molecules that have similar structures.7 Each member of this group has a cobalt atom surrounded by a macrocyclic ligand (a corrin ring) that bears various substituents. In addition to the corrin ring the cobalt atom in each of these compounds also is coordinated with a ligand that contains a phosphate group, a sugar moiety, and a nitrogenous base. Compounds related to 5 differ from each other in the structure of the R group attached to cobalt. R represents the 5'-deoxyadenosyl group in coenzyme B12 (5), but for related compounds R can be as structurally simple as a methyl or hydroxyl group.7
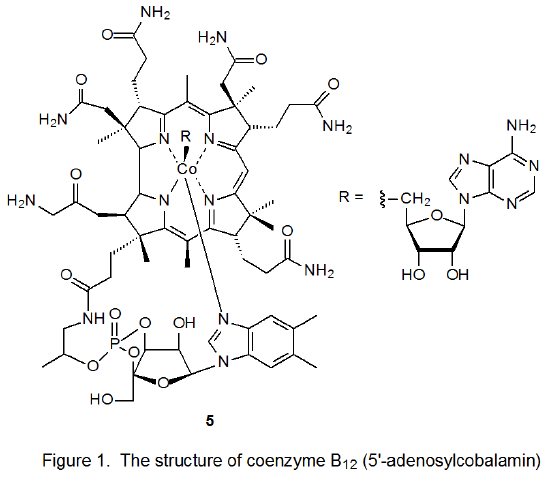
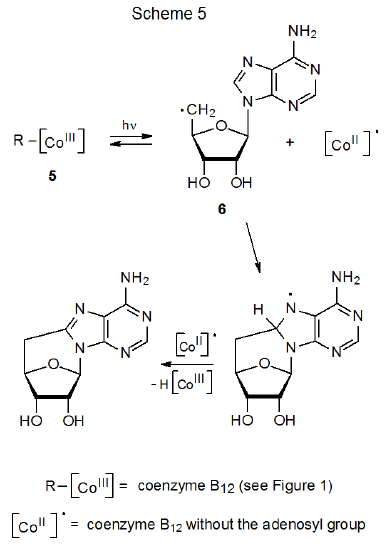
The original stimulus for study of carbon–cobalt bond homolysis as a pathway for forming carbon-centered radicals came from investigation of the reactions of coenzyme B12 (5).8–10 In biological systems enzyme-induced homolysis of the carbon–cobalt bond in 5 produces the 5'-deoxyadenosyl radical 6 and the cobalt-centered radical 7 (B12r, eq 1).8–10 In experiments outside biological settings the 5'-deoxyadenosyl radical (6) is produced from coenzyme B12 (5) by photolysis with visible light.11 When photolysis is conducted in the absence of an effective hydrogen-atom donor or other radical trap, cyclization follows homolysis of the carbon–cobalt bond (Scheme 5).8-10,12
.png?revision=1&size=bestfit&width=430&height=213)
b. Cobaloxime Complexes
The discovery that carbon–cobalt bond homolysis in coenzyme B12 (5) produced the carbon-centered radical 6 (eq 1), led to investigation of simpler molecules that could model this behavior. Cobaloximes are one of several types of compounds found to be effective choices for this role.13–16 Carbohydrate cobaloximes 8 and 9 produce radicals 10 and 4, which recombine in the absence of radical traps (Scheme 6).13 In the presence of compounds that react with radicals, 10 and 4 undergo characteristic radical reactions; thus, the D-mannopyranos-1-yl radical 10 adds to acrylonitrile (11) to give the adduct radical 12, which then combines with ·Co(dmgH)2py (4) to form the addition product 13 (Scheme 7).13
.png?revision=1){kind=link}
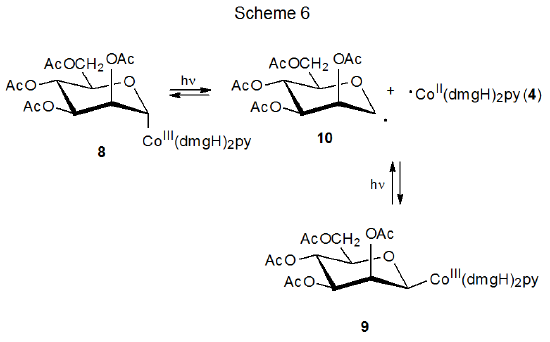
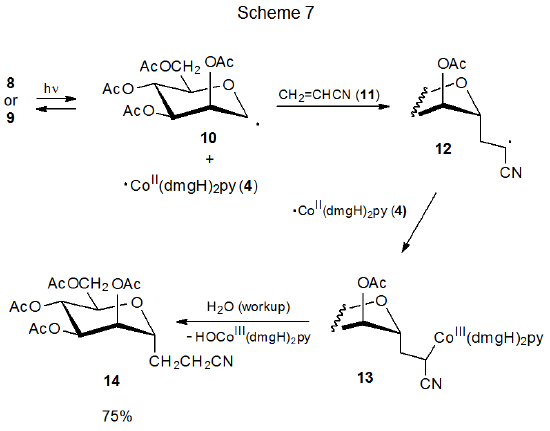
A necessary condition for the reaction shown in Scheme 7 is that 4 [Co(dmgH)2py] be stable enough to remain unchanged while the addition of 10 to 11 is taking place. The needed stability of 4 derives from protection of its radical center by the attached ligands; thus, 4 can be viewed as a persistent radical.
c. The Persistent-Radical Effect
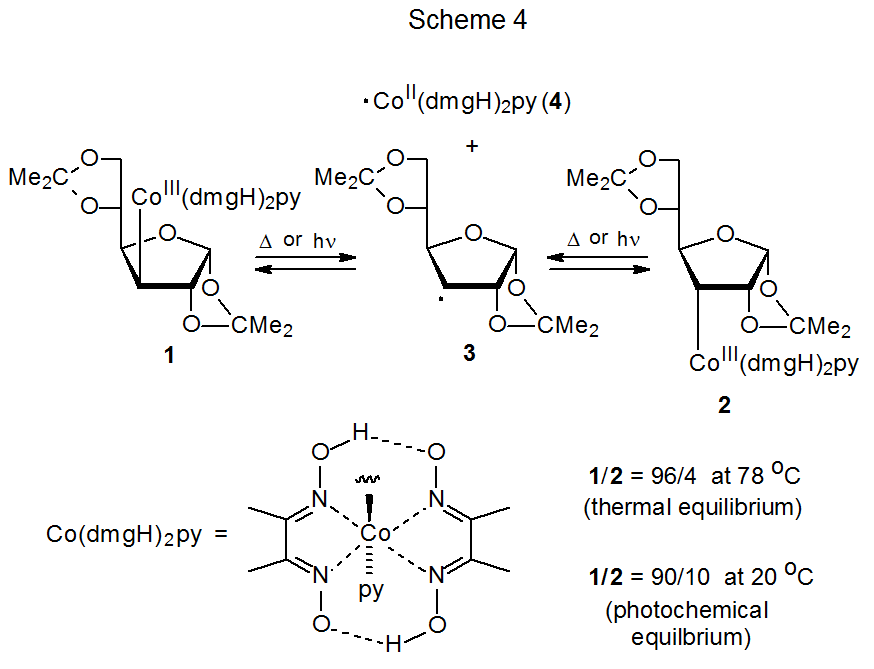
Persistent radicals, such as ·Co(dmgH)2py (4), are responsible for a type of reactivity known as the persistent-radical effect.17–19 This effect causes a reaction that generates a persistent radical (R1·) and a transient radical (R2·) in equal amounts to give a higher yield of the cross-coupling product (R1R2) than would be expected from random radical coupling. The explanation for greater cross-coupling product formation begins with the recognition that although persistent and transient radicals are formed in equal amounts, this equality is short lived. Due to the reactive nature of transient radicals their concentration decreases more rapidly in the early stages of a reaction than does the concentration of persistent radicals. (Transient radicals combine, disproportionate, and undergo other reactions much more rapidly than persistent radicals.) The rapidly developed, higher concentration of persistent radicals in the early stages of reaction means that any newly formed, transient radical is more likely to encounter and combine with a persistent radical than with another transient one; in other words, the cross-coupling product R1R2 becomes the major coupling product.
An example of the persistent radical effect is shown in the reaction given in Scheme 4, where carbon–cobalt bond homolysis in 1 or 2 produces the persistent radical 4 and the transient radical 3. Even with the extended heating or photolysis needed to reach equilibrium, there was no evidence of formation of a coupling product other than the cross-coupling products 1 and 2. The persistent radical effect also is operative in the addition reaction shown in Scheme 7. In this case the transient radical 12, produced by addition of 10 to acrylonitrile (11) , and the persistent radical 4 combine to form the only radical-coupling product isolated.
{kind=link}
{kind=link}
2. Carbon-Mercury Bond Homolysis
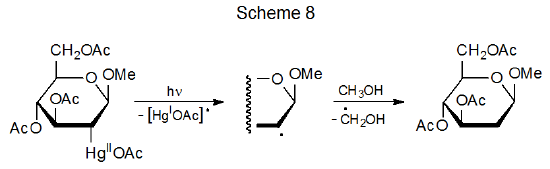
There are similarities in reactivity among compounds with C–Co and C–Hg bonds. Both bonds are strong enough to exist in stable structures at room temperature but both readily cleave upon photolysis. The result in each case is formation of a metal-centered and a carbon-centered radical. Carbon-centered radicals produced by carbon–mercury bond homolysis undergo typical radical reactions, such as the hydrogen-atom abstraction shown in Scheme 8.20
3. Manganese(III) Acetate [Mn(OAc)3] Reactions
Carbon-centered radicals can be generated by reaction of manganese(III) acetate with CH-acidic compounds such as the β-diketone shown in Scheme 9.21–24 The first step in this process is formation of the enolate 15.23 In the presence of an unsaturated compound two mechanisms for reaction of 15 are considered to be possible. In the first of these electron transfer forms manganese(II) acetate and the resonance-stabilized radical 16, which then adds to an unsaturated compound. A second possible pathway for addition is a concerted process in which the enolate 15 reacts directly with the unsaturated compound to produce the adduct radical 17 (Scheme 9).23 Reaction by either of these pathways is believed to take place by inner-sphere electron transfer.
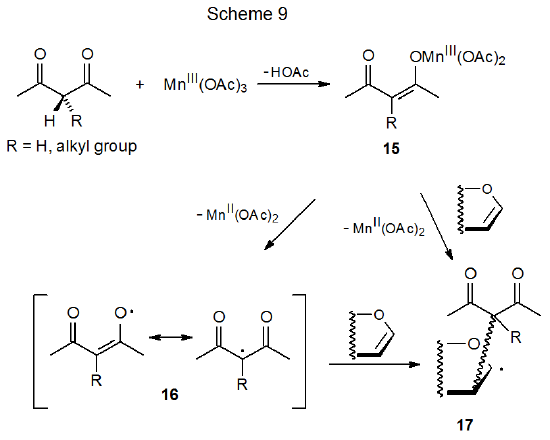
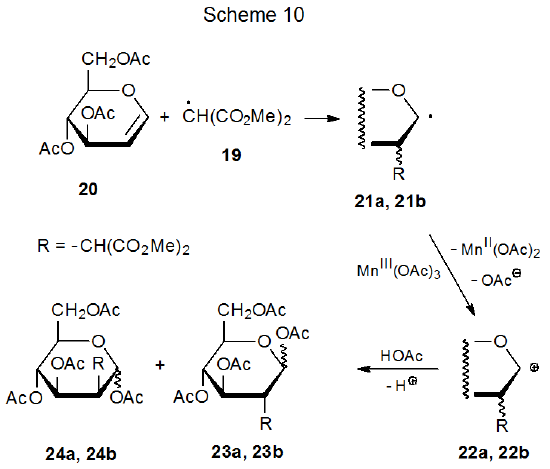
Since radical centers with two, attached carbonyl groups are electrophilic, radicals such as 16 (Scheme 9) add most easily to unsaturated compounds with electron-rich multiple bonds.22 This is the point at which carbohydrates typically become involved in reactions begun by manganese(III) acetate because glycals have electron-rich π systems that are attractive targets for addition of electrophilic radicals; for example, the radical 19, formed by reaction of dimethylmalonate (18) with manganese(III) acetate (eq 2), adds to the tri-O-acetyl-D-glucal 20 to produce the stereoisomeric radicals 21a and 21b (Scheme 10).25,26 This addition, which occurs regioselectively at C-2, is followed by oxidation of the resulting radicals with a second molecule of manganese(III) acetate to give the corresponding cations 22a and 22b. These cations react with the solvent (acetic acid) to yield the final products (23a, 23b, 24a, and 24b). Manganese(III) acetate, therefore, is involved in both the formation and disappearance of the radicals in this reaction. (Electrophilic radicals and other aspects of radical philicity are discussed in Chapter 7.)
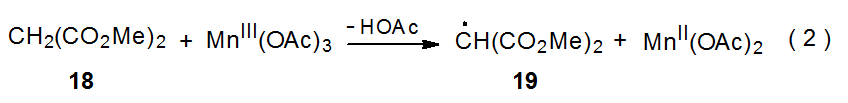.png?revision=1&size=bestfit&width=425&height=52)
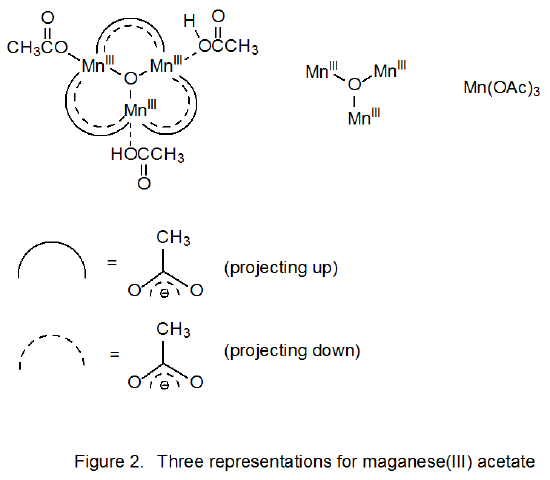
Manganese(III) acetate has a more complicated structure than the formula Mn(OAc)3 indicates. It is an oxo-centered trimer of three manganese ions held together by six bridging acetates.27–29 Three representations for this structure are shown in Figure 2. It is often convenient in discussing reactions of this compound to use the abbreviated formula Mn(OAc)3.
4. Ammonium Cerium(IV) Nitrate [(NH4)2Ce(NO3)6] Reactions
Reaction of CH-acidic compounds with ammonium cerium(IV) nitrate generates electrophilic, resonance-stabilized radicals in a manner similar to reaction with manganese(III) acetate.30,31 As mentioned in the previous section, these radicals add readily to the electron-rich double bonds such those found in glycals (eq 3).30 Oxidation of CH-acidic compounds with ammonium cerium(IV) nitrate to produce electrophilic radicals has the advantage, when compared to reactions with manganese(III) acetate, of being able to be conducted at or below room temperature. [The reactions of manganese(III) acetate and ammonium cerium(IV) nitrate are discussed further in Chapter 21 of Volume II.]
.png?revision=1&size=bestfit&width=435&height=171)