II. Phosphatoxy Group Migration
- Page ID
- 24049
A. Reaction Mechanism
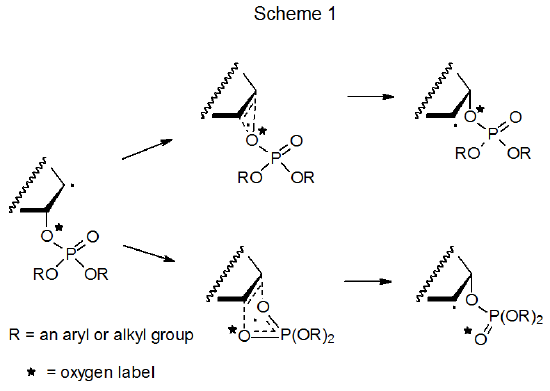
Initially two mechanisms were considered as possibilities for phosphatoxy group migration of the type shown in eq 1. (This same mechanistic choice exists for acyloxy group migration and is discussed in Section V.A of Chapter 8.) The first of these mechanisms consisted of a pair of competing, concerted reactions, each of which passed through a cyclic transition state (Scheme 1).3–7 A basic difference between this pair was that in one reaction the same oxygen atom was bonded to the carbon-atom framework both before and after migration, but in the other the framework had a different oxygen atom attached after migration. Proposing migration via a combination of these two reactions made it possible to explain experiments with oxygen-labeled substrates in which only a portion of the labeled oxygen was attached to the carbon-atom framework after migration. The results from early studies favored this two-reaction explanation,3–7 but those from later investigations required it to be changed because the later studies showed that ionic intermediates were involved in the migration process.
.png?revision=1){kind=link}
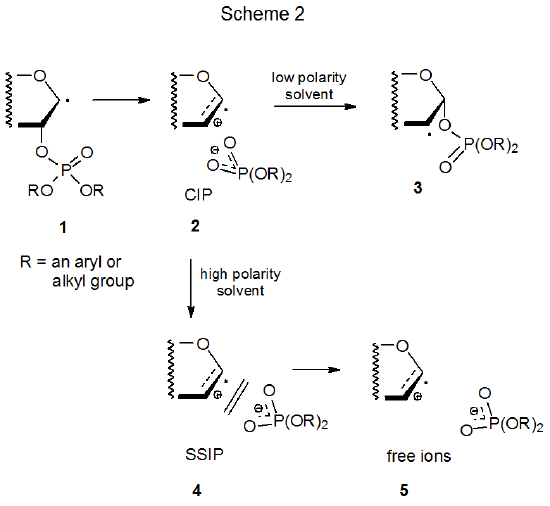
A mechanism that satisfies the ionic-intermediate requirement is shown in Scheme 2, where the β-phosphatoxy radical 1 fragments heterolytically to give the contact ion pair (CIP) 2.8–13 This ion pair recombines in low polarity solvents to form the group-migrated radical 3, but in more polar solvents the CIP also can separate to become a solvent-separated ion pair (SSIP, 4) and then free ions 5.10
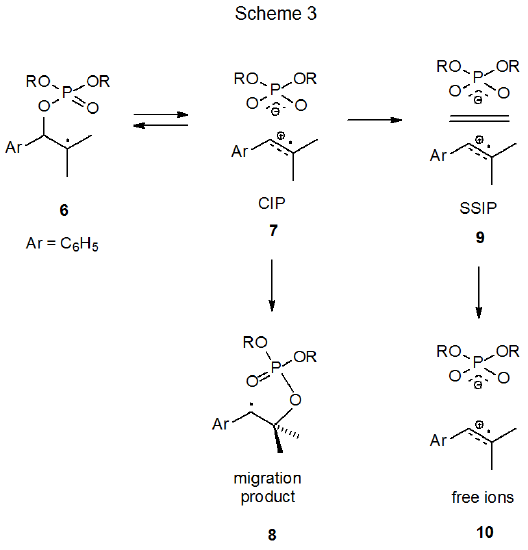
Critical support for the ion-pair mechanism for phosphatoxy group migration comes from laser-flash-photolysis (LFP) experiments. Both the SSIP 9 and the diffusively free radical cation 10 can be detected in studies where LFP generates the radical 6 (Scheme 3).12 Evidence for the CIP in this reaction is indirect presumably because its lifetime is too short to permit direct detection. Study of reaction rates in solvents of different polarity supports the idea that the radical 6 is passing through a common intermediate in forming either the migrated radical 8 or the SSIP 9. A reasonable conclusion is that the common intermediate is the contact ion pair 7 (Scheme 3).12 Entropies of activation, which are the same for ion-pair formation in high polarity solvents and group migration in solvents of low polarity, also favor a common intermediate for which 7 is the prime candidate.10,12 Generalizing these results leads to the reaction mechanism proposed in Scheme 2. (The wording in this paragraph also is found in Section V.A.5 of Chapter 8 because the information contained is pertinent to the mechanism of acyloxy group migration.)
Phosphatoxy group migrations are not wide-spread in carbohydrate chemistry; in fact, all reported reactions involve migration from C-2 to C-1 in a pyranoid or furanoid ring. This situation exists because the stabilization afforded by the ring oxygen atom is critical at the transition state leading to radical-cation formation. Examining the reactivity of the noncarbohydrate radicals shown in equations 4 and 5 is instructive. An oxygen atom must be fully able to participate in radical-cation stabilization for heterolytic bond breaking to occur (eq 4).14 Replacing the methoxy group in the substrate in the reaction shown in eq 4 with an acetyl group, as is done in the reaction shown in eq 5, prevents radical-cation formation because an oxygen atom with an electron-withdrawing group attached is unable to stabilize sufficiently the transition state leading to the radical-cation intermediate.14
.png?revision=1&size=bestfit&width=285&height=111)
.png?revision=1&size=bestfit&width=285&height=116)
B. Relative Reaction Rates
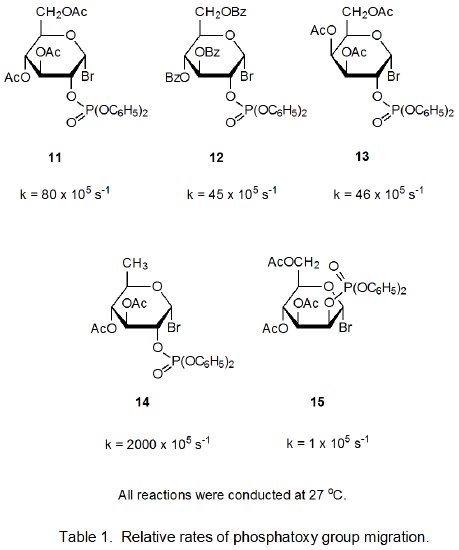
Relative rate constants for phosphatoxy group migration in the reactions of the five hexopyranosyl bromides 11-15 are given in Table 1.15 The rate constant for reaction of the 6-deoxy bromide 14 is substantially larger than those for the other bromides. Replacing the electron-withdrawing acyloxy group at C-6 with a hydrogen atom makes the radical cation 16 more stable and, in so doing, stabilizes the transition state leading to it (eq 6).
.png?revision=1&size=bestfit&width=325&height=164)
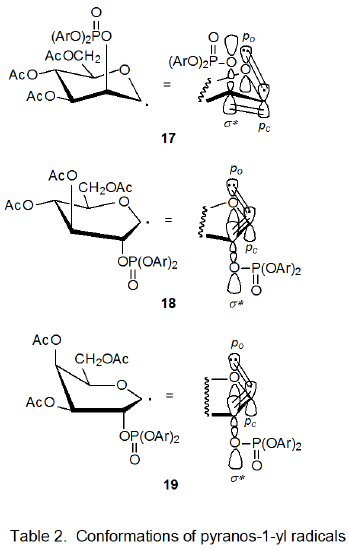
The rate constant for reaction of the D-mannopyranosyl bromide 15 is decidedly smaller than those for reactions of the bromides 11-14. One factor that contributes to this reduced reactivity is the enhanced stability of the radical 17 when compared to the corresponding radicals derived from the other bromides (11-14). Only 17 remains in a relatively strain-free, 4C1 conformation while taking advantage of the stabilizing interaction of parallel po, pc, and σ* orbitals (Table 2).15 To benefit from parallel-orbital stabilization, the radicals derived from bromides 11-14 must assume less stable conformations; for example, the radical derived from 11 adopts the B2,5 boat conformation 18.15 As migration takes place in each of the radicals 17-19, po, pc, σ* orbital stabilization is lost, but for radicals 18 and 19 this loss is compensated for, at least in part, by movement toward a more stable, 4C1 conformation. Such compensation means that the transition states for radical-cation formation from 18 and 19 are not as high in energy as that for reaction of 17; consequently, group migration for the radical 17 is slower than for 18 and 19.
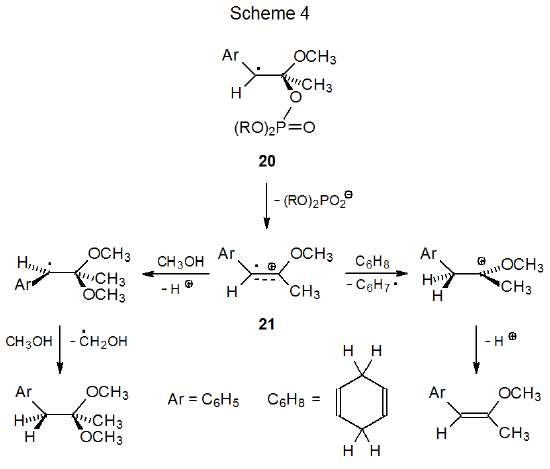
Pertinent information about formation and reactivity of radical cations comes from the study of noncarbohydrate systems.16,17 Nucleophilic trapping of the radical cation 21 by methanol (k< 1 x 103 M-1s-1) is slow compared to hydrogen-atom abstraction from 1,4-cyclohexadiene (k = 6 x 105 M-1s-1) (Scheme 4).17 To the extent that this observation is a general one, radical cations can be expected to have greater radical reactivity than cationic reactivity.
C. Stereoselectivity
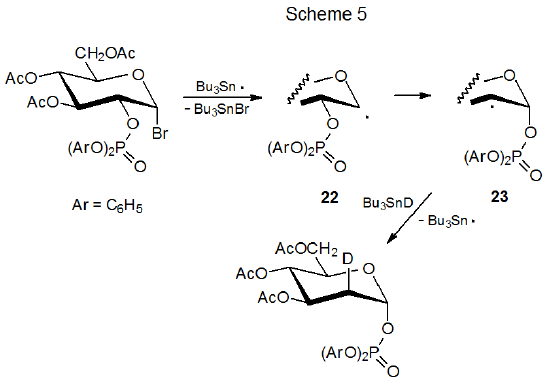
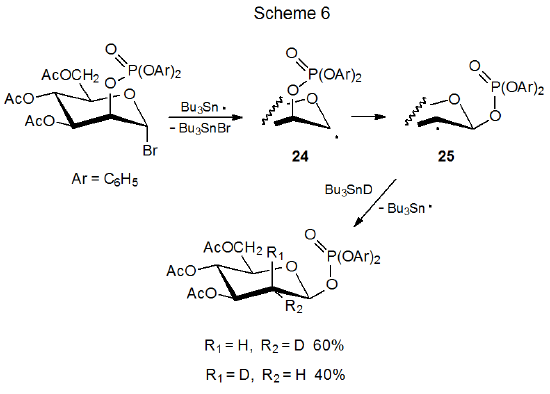
The reactions pictured in Schemes 5 and 6 show that phosphatoxy group migration is a stereospecific process; thus, the epimeric radicals 22 and 24 give the product radicals 23 and 25, respectively.1,15 Once migration has taken place, stereoselective deuterium abstraction completes the reaction. For the radical 23 abstraction is highly stereoselective, but it is much less so for the radical 25. Shielding of the α face of 23 by the axial phosphatoxy group causes deuterium to be abstracted from the β face of this radical (Scheme 5). The equatorial phosphatoxy group in 25 is not nearly as effective at forcing Bu3SnD to the opposite face of the pyranoid ring (Scheme 6).
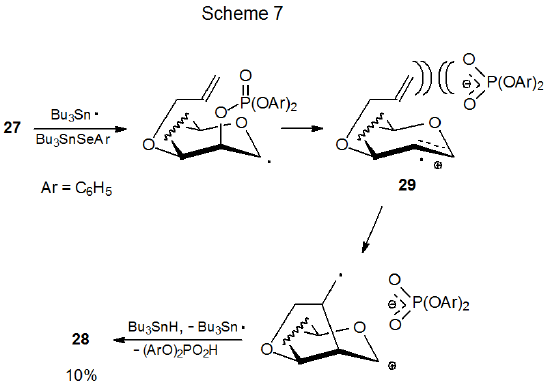
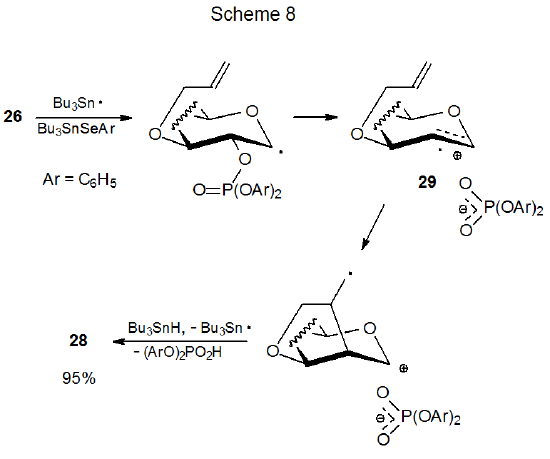
Differential shielding of the faces of a pyranoid ring affects the ability of the phosphates 26 and 27 to undergo new ring formation (eq 7). The phosphate 26 gives a decidedly higher yield of the glycal 28 than does its epimer 27.18 The substantially lower product yield from reaction of 27 is attributed to steric hindrance by the nearby phosphate counter ion during cyclization of the radical cation 29 (Scheme 7). When 29 is generated from 26, however, the counter ion is on the opposite face of the ring and does not impede cyclization (Scheme 8).
.png?revision=1&size=bestfit&width=435&height=200)