
VII. Factors Affecting O-Thiocarbonyl Compound Synthesis
- Page ID
- 24069
A. The Strength of the Participating Nucleophile
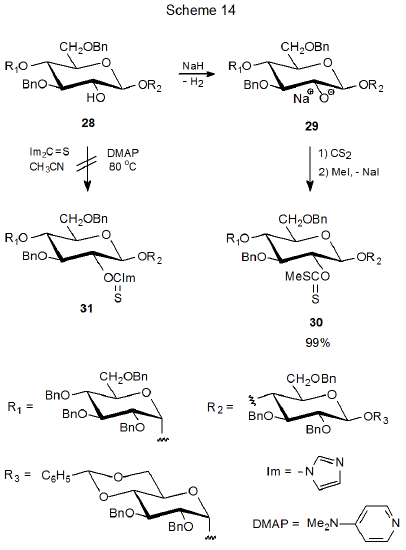
Some compounds do not form every type of O-thiocarbonyl derivative. The tetrasaccharide 28, for example, does not produce a (thiocarbonyl)imidazolide (31) but does form a xanthate (30) (Scheme 14).62 A possible explanation for this difference in behavior is based upon the reactivity of the nucleophiles involved in preparation of each derivative. The first step in xanthate formation is conversion of 28 into the powerful nucleophile 29 by deprotonation of the C-2' hydroxyl group with sodium hydride. These reaction conditions stand in contrast to those for (thiocarbonyl)imidazolide synthesis, which depends upon the less effective nucleophile 28. (The small equilibrium concentration of the alkoxide ion 29, produced by DMAP deprotonation of 28, is insufficient to cause the (thiocarbonyl)imidazolide 31 to form in detectable amounts).
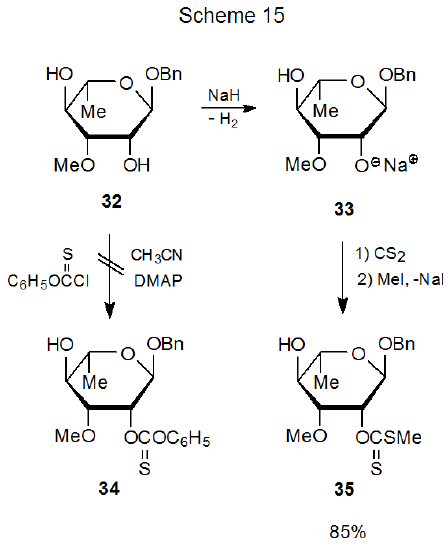
Another example illustrating the role of nucleophilicity in producing O‑thiocarbonyl compounds concerns the phenoxythionocarbonate 34, which cannot be prepared from the diol 32, even though the xanthate 35 easily forms from this compound (32) by way of the alkoxide ion 33 (Scheme 15).63 Once again, greater ease in xanthate formation can be linked to greater nucleophilicity of an alkoxide ion when compared to its corresponding alcohol.
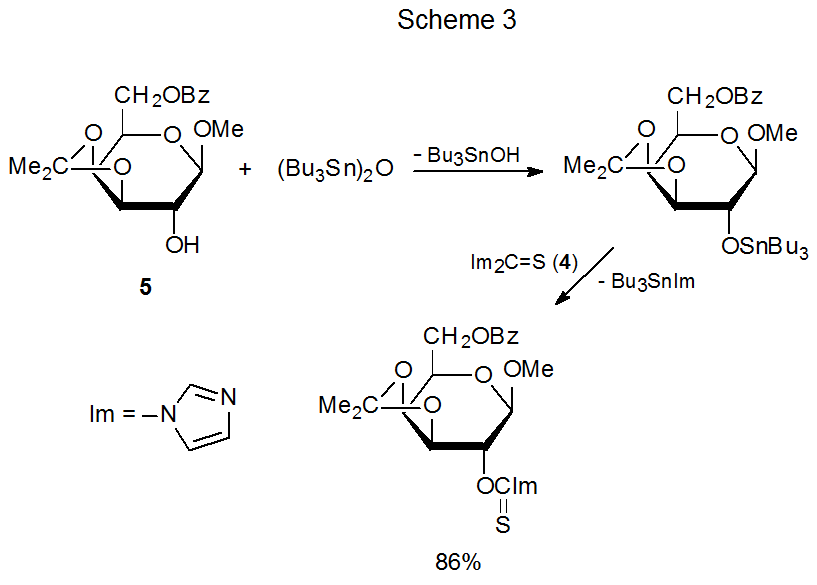
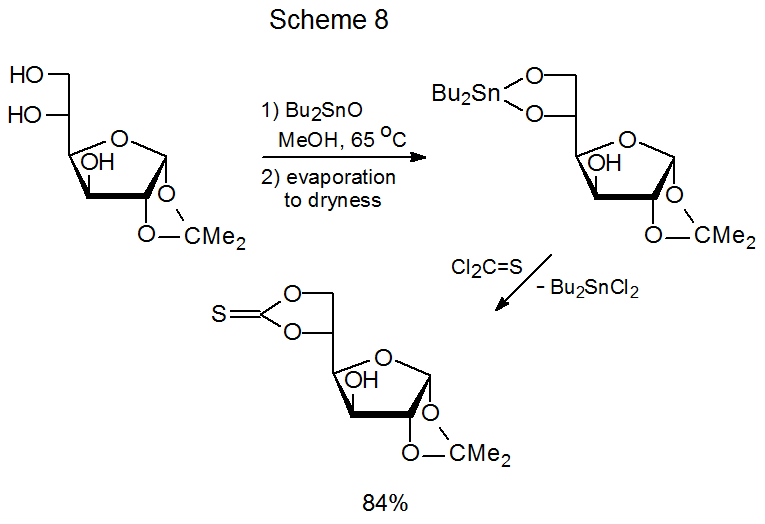
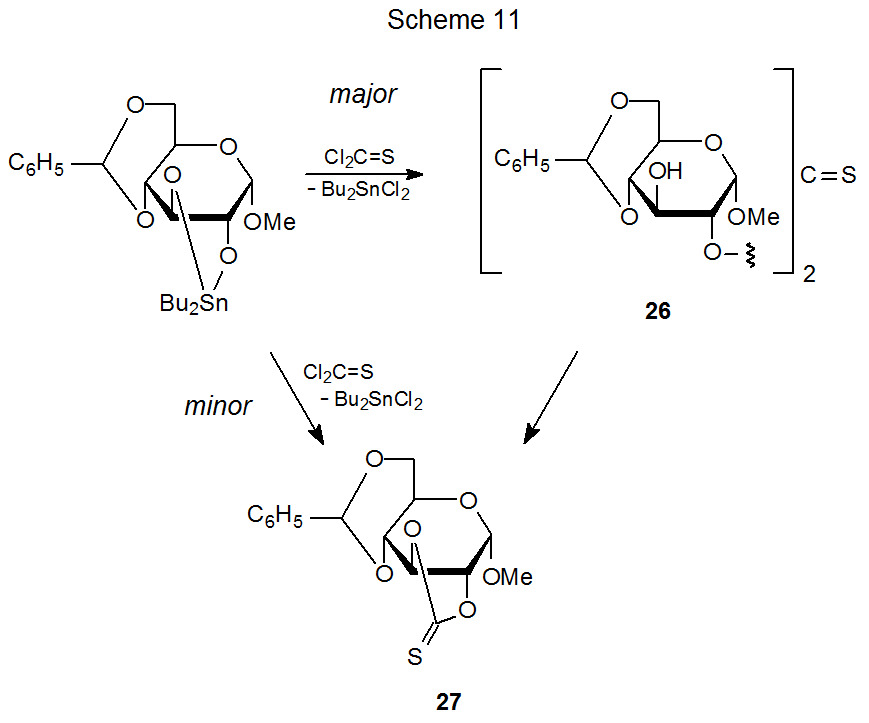
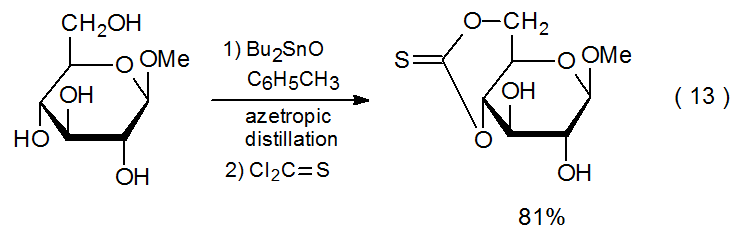
A method for increasing the nucleophilicity of a partially protected carbohydrate without converting it into a fully ionic compound consists of forming a derivative containing a tin–oxygen bond. This is the approach adopted in several of the reactions (Schemes 2, 3, 8, and 11 and equations 12 and 13) discussed thus far. In the derivatization shown in Scheme 3, for example, combining the methyl glycoside 5 with bis(tributyltin)oxide forms a nucleophile able to produce a (thiocarbonyl)imidazolide, but reaction of 5 without increasing its nucleophilicity is unsuccessful.20
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.png?revision=1){kind=link}
.png?revision=1){kind=link}
Another advantage of the nucleophilicity of an alkoxide ion when participating in xanthate synthesis is that reaction can take place at low temperatures.64,65 Reaction occurring under these conditions is particularly important for forming tertiary xanthates (eq 1564) because these compounds readily undergo thermal rearrangement and elimination reactions.66
.png?revision=1&size=bestfit&width=360&height=102)
B. Protecting-Group Migration and Loss
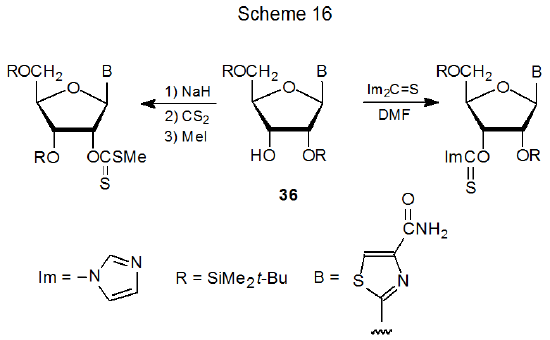
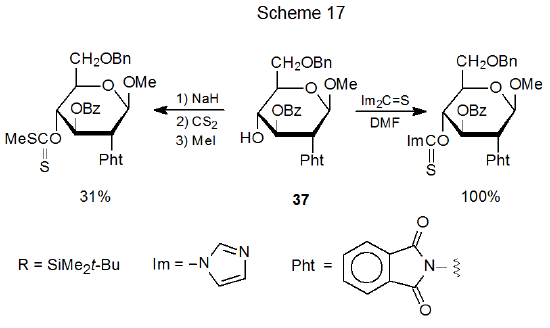
Although increasing the nucleophilicity of a hydroxyl group by deprotonation is sometimes helpful in forming an O-thiocarbonyl compound, deprotonation also promotes protecting group migration. Compound 36, for example, forms a (thiocarbonyl)imidazolide with the silyl group remaining in place, but attempted synthesis of the corresponding xanthate causes complete O-2' to O-3' silyl-group migration (Scheme 16).67 In another example, compound 37 forms a xanthate in only 31% yield, but (thiocarbonyl)imidazolide formation is quantitative (Scheme 17).68 Group migration (Scheme 16) and reduced product yield (Scheme 17) (possibly through benzoyl group loss or migration or both) are linked to the nucleophilicity of the alkoxide ions formed during xanthate synthesis.
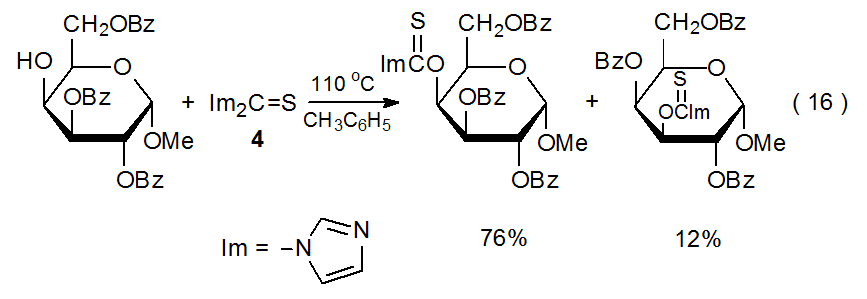
Even though the absence of a strong base during (thiocarbonyl)imidazolide formation reduces the likelihood of group migration, it does not eliminate this possibility entirely. Whenever a carbon atom bearing a hydroxyl group has an acyloxy or silyloxy group on a neighboring (or nearby) atom, group migration is a possibility15,69,70 because the organic base (and catalyst) imidazole is generated as the reaction proceeds (eq 2). An example of a migration reaction that takes place during (thiocarbonyl)imidazolide synthesis is shown in eq 16, where the benzoyl group at O-3 in the starting material migrates to O-4 in forming the minor product.15
.png?revision=1){kind=link}
.png?revision=1&size=bestfit&width=435&height=150)
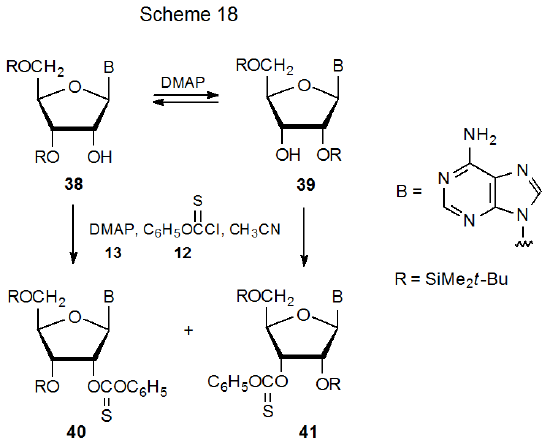
The possibility that imidazole causes group migration during (thiocarbonyl)imidazolide formation garners support from the observation that DMAP causes such reaction during thionocarbonate synthesis. Phenyl thionocarbonates 40 and 41 both form when either nucleoside 38 or 39 reacts with phenoxythiocarbonyl chloride (12) (Scheme 18).71 The formation of this mixture of products (40 and 41) is the result of DMAP-catalyzed, silyl-group migration in compounds 38 and 39 prior to esterification (Scheme 18).
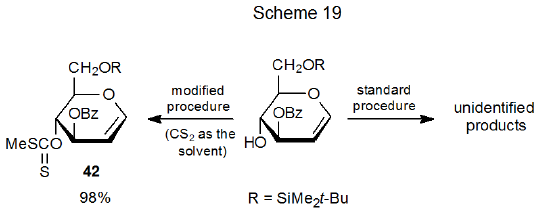
Group migration sometimes can be avoided by modification in the reaction conditions. The xanthate 42, for instance, cannot be synthesized by the standard procedure, but it forms in excellent yield when carbon disulfide is the reaction solvent (Scheme 19).72 When carbon disulfide is present in large excess, the increased rate of xanthate formation suppresses competing, unimolecular reactions such as group migration.
C. Displacement Reactions
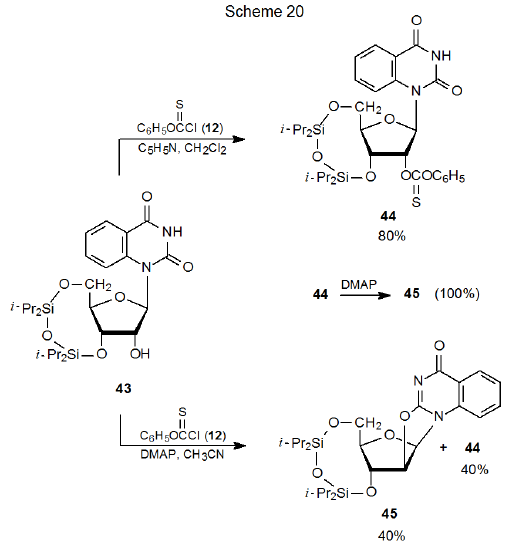
O-Thiocarbonyl groups can function as nucleofuges in displacement reactions. They are not particularly effective in this role; consequently, their participation is limited to internal reaction in which the nucleophile is created by deprotonation and is held in an advantageous position for reaction. An example of internal displacement of this type is shown in Scheme 20 where the thionocarbonate 44 forms in good yield from reaction of the nucleoside 43 with phenoxythiocarbonyl chloride (12) in the presence of pyridine, but when the stronger base DMAP (13) is used, internal SN2 displacement produces the anhydro nucleoside 45.73 Support for the idea that 44 is an intermediate in this reaction comes from its quantitative conversion into 45 by reaction with DMAP.
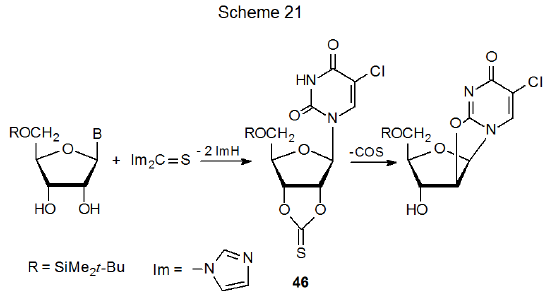
Cyclic thionocarbonates also can be substrates in nucleophilic substitution reactions.74,75 In the reaction shown in Scheme 21, for example, formation of the 2',3'-O-thiocarbonyl derivative 46 places nucleofuges at C-2' and C-3'. The C-2' substituent then is displaced by an oxygen atom in the nitrogenous base portion of the molecule.74
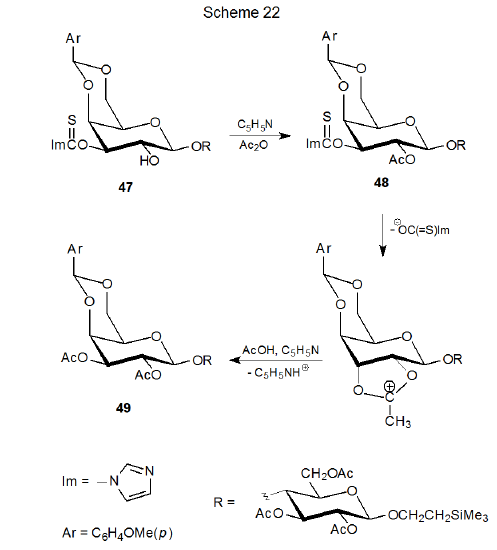
Another example of nucleophilic substitution involving an O-thiocarbonyl compound is found in Scheme 22, where attempted acetylation of the disaccharide 47 causes replacement of the O-imidazol-1-ylthiocarbonyl group with an acetyl group.10 A reasonable assumption is that the desired acetate 48 actually forms, but the O‑imidazol-1-ylthiocarbonyl group is a sufficiently good nucleofuge that it is displaced by the neighboring O-acetyl group in a reaction that leads to the pentaacetate 49. Acetylation of the closely related xanthate 50 (eq 17) without internal displacement indicates that the O-[(phenylthio)thiocarbonyl] group is a less effective nucleofuge.10
.png?revision=1&size=bestfit&width=425&height=186)
D. Regioselective Reactions
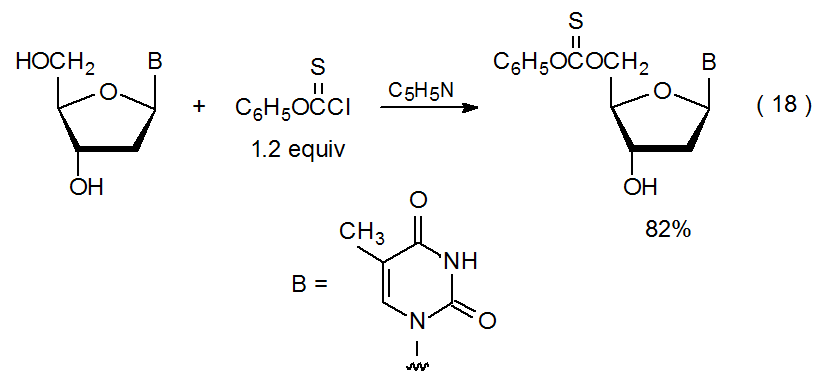
In a carbohydrate with more than one unprotected hydroxyl group, it is sometimes possible to predict which group will react preferentially with a limited amount of a thioacylating agent. For example, reaction of the less hindered of two hydroxyl groups will occur if there is a substantial difference in their steric shielding; thus, in the reaction shown in eq 18, regioselective thioacylation takes place at the primary, rather than the secondary, hydroxyl group.76
.png?revision=1&size=bestfit&width=415&height=190)
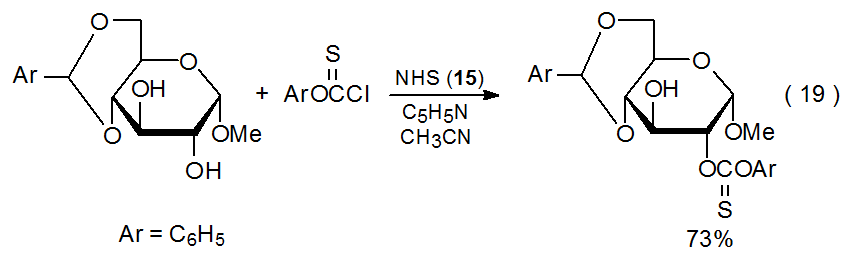
Even if there is little difference in steric shielding of two hydroxyl groups, site selectivity sometimes can be predicted if one of the groups is attached to C-2 and deprotonation is the first step in the reaction. Under these conditions the typically greater acidity of the C-2 hydroxyl group determines which of the two possible alkoxide ions will form to a greater extent. This preferential formation leads to regioselective reaction at C-2 (Scheme 1563 and eq 1930). Comparing the reaction shown in eq 1930 with that in eq 141 demonstrates that predicting regioselective reaction at C-2 must be done cautiously. In these two reactions the same compound exhibits different selectivity when the reagents and the reaction conditions change.
{kind=link}
.png?revision=1){kind=link}
.png?revision=1&size=bestfit&width=430&height=132)
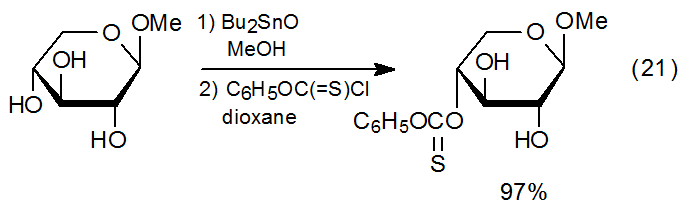
Regioselectivity extends to reactions where esterification is preceded by formation of a stannylene complex (equations 12 and 13). Since this selectivity is dependent upon the stability and reactivity of the various stannylene complexes that are in equilibrium in the reaction mixture, predicting or even rationalizing the formation of reaction products is complicated by esterification being a two-step process with selectivity involved in each step. Although regioselective reaction of stannylene complexes is often high, it is far from assured, as is illustrated by the nearly unselective reaction of the methyl glycoside 51 (eq 20).52 The difficulty in predicting site selectivity is underscored when comparing the reaction shown in eq 20 with that in eq 21, where an essentially unselective reaction becomes highly selective upon changing the configuration of the methoxy group at C-1.52 Under carefully selected conditions reaction of organotin complexes of a variety of unprotected methyl glycosides with phenoxythiocarbonyl chloride leads to highly regioselective thionocarbonate formation.53
.png?revision=1&size=bestfit&width=435&height=109)
.png?revision=1&size=bestfit&width=345&height=107)