II. Deoxygenation: The Barton-McCombie Reaction
- Page ID
- 24072
A. A Two-Step Sequence
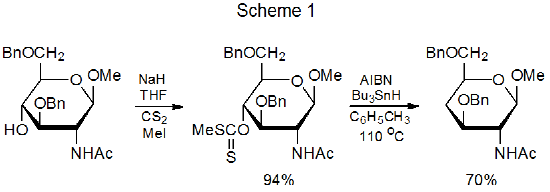
In 1975 Barton and McCombie reported a two-step sequence for hydroxyl-group replacement by a hydrogen atom.1 The first step in this process was the conversion of the hydroxyl group into an O-thiocarbonyl group, and the second step (the Barton-McCombie reaction) was a free-radical chain reaction that replaced the O-thiocarbonyl group with a hydrogen atom. A typical example of this widely used, reaction sequence is shown in Scheme 1.2
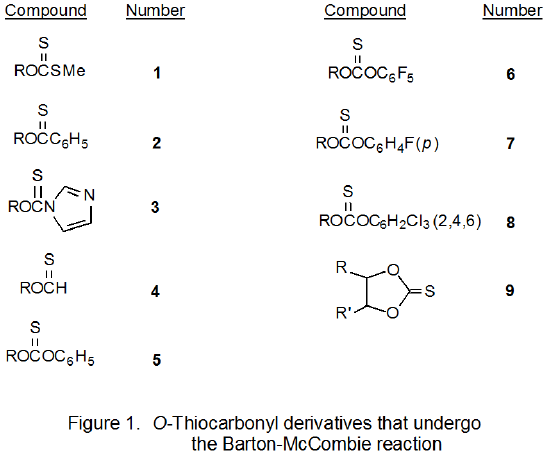
Various types of O-thiocarbonyl compounds undergo the Barton-McCombie reaction. Initially this group consisted of xanthates (1), thionobenzoates (2), thiocarbonylimidazolides (3), and thionoformates (4) (Figure 1).1 Subsequently, this list was expanded to contain phenyl thionocarbonates (5),3,4 including those with electron-withdrawing substituents in the aromatic ring (6-8),5,6 and cyclic thionocarbonates (9).7,8
B. Proposed Reaction Mechanisms
A proposed mechanism for the Barton-McCombie reaction is shown in Scheme 2.1,9 In the initiation phase of this reaction thermal decomposition of 2,2'-azobis(isobutyronitrile) (AIBN) (eq 1), the most common initiator for the Barton-McCombie reaction, produces a radical that abstracts a hydrogen atom from tri-n-butyltin hydride (eq 2). In the first propagation step the tri-n-butyltin radical adds to a carbon–sulfur double bond to create the adduct radical 10 (eq 3). Reaction reaches a critical stage at this point because its success requires 10 to fragment to give the radical 11 (eq 4) before competing reactions can intervene. Once fragmentation takes place, hydrogen-atom abstraction by 11 from tri-n-butyltin hydride completes the overall reaction and generates a new, chain- carrying, tri-n-butyltin radical (eq 5). (Equations 1-5 are found in Scheme 2.)
.png?revision=1&size=bestfit&width=400&height=632)
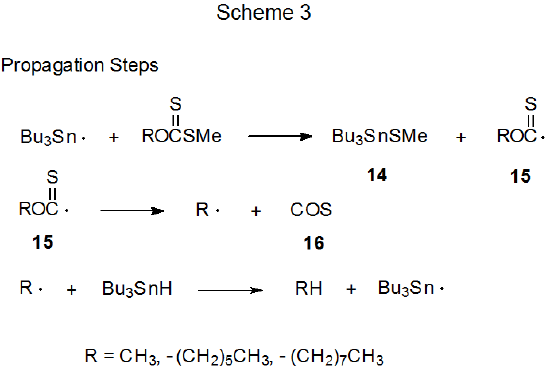
The propagation steps for a revised mechanistic proposal for the Barton-McCombie reaction are shown in Scheme 3.10 (The initiation and termination steps for this mechanism are the same as those pictured in Scheme 2.) The primary change introduced in the revised mechanism (Scheme 3) is that Bu3Sn· does not add to the thiocarbonyl group but rather abstracts the SCH3 group. Identification of the radical 15 in the ESR spectrum of the reaction mixture supports the revised mechanism; however, an argument against mechanistic significance of 15 is that this intermediate is observed under conditions quite different from those of the Barton-McCombie reaction (e.g., no effective hydrogen-atom donor (Bu3SnH) was present in the reaction mixture because the tri-n-butyltin radicals were generated from photolysis of Bu3SnSnBu3.10)
Subsequent competition experiments returned support to the original mechanism (Scheme 2).11–13 In addition to these experiments, 119Sn NMR identification of the intermediate 12 (R = SCH3) in a Barton-McCombie reaction mixture provided evidence for the addition of Bu3Sn· to the thiocarbonyl group, as proposed in Scheme 2, rather than abstraction of SCH3, as proposed in Scheme 3.13 Further mechanistic analysis led to the conclusion that the tri-n-butyltin radical must be adding reversibly to the thiocarbonyl group to give the radical 10, which then fragments as shown in eq 4 (Scheme 2). Additional support for this mechanism is based on a ring-forming reaction that is described at the end of this chapter, after cyclization reactions have been discussed.
.png?revision=1){kind=link}
C. Hydrogen-Atom Donors/Chain-Transfer Agents
Critical to the success of the Barton-McCombie reaction is the compound that donates a hydrogen atom to the carbohydrate radical (eq 5) to complete the propagation sequence. Tri-n- butyltin hydride is particularly well suited for this role because it rapidly donates a hydrogen atom to a carbon-centered radical, and in the same reaction generates the chain-carrying radical Bu3Sn·, an intermediate needed to begin a new sequence of propagation steps (eq 3). (A critical feature of the reactivity of Bu3Sn· is that it does not cause side reactions by abstracting hydrogen atoms from carbon-hydrogen bonds.)
Although use of tri-n-butyltin hydride has significant advantages, it also suffers from substantial drawbacks. There are serious problems associated with the toxicity of tin-containing compounds and the difficulty in removing residues of these compounds from reaction products. A variety of solutions to these problems have been proposed. Because these solutions apply not just to O-thiocarbonyl compounds but also to a broad range of carbohydrate derivatives, they will not be discussed here; rather, they have been gathered together and are found in Appendix I.
D. The Scope and Reactivity of O-Thiocarbonyl Compounds
The number of O-thiocarbonyl carbohydrate derivatives that undergo the Barton-McCombie reaction is large and continues to grow. Although all of these compounds react basically in the same way, some are better suited than others for particular situations. The following several sections focus on the scope and special reactivity of various O-thiocarbonyl compounds. To emphasize their broad range of reactivity, references are provided to Barton-McCombie reaction taking place at various positions in substituted carbohydrates. These references are not meant to represent the total number that exists, but rather to provide examples of the reactions possible in cyclic and open-chain carbohydrates.
1. Xanthates
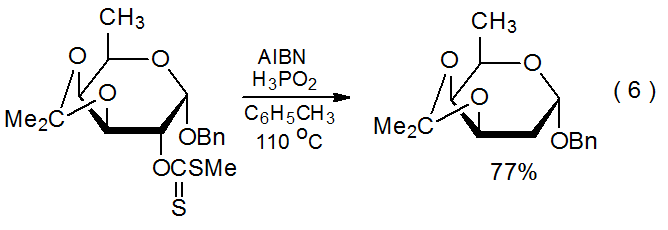
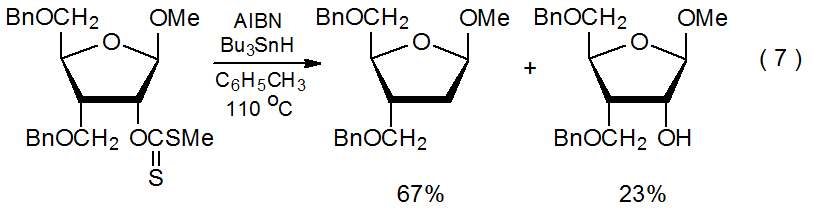
A striking feature of xanthate reactivity is the number and variety of carbohydrates that can be deoxygenated by xanthate formation followed by Barton-McCombie reaction. This sequence replaces hydroxyl groups at the 2-,14,15 3-,16,17 4-,2,18 and 6-19,20 positions with hydrogen atoms in compounds containing pyranoid rings, as well as the 1-,21 2-,22,23 3-,24,25 5-,26 and 6-27positions in those with furanoid rings. Equations 615 and 728 provide typical examples of reactions of carbohydrates containing pyranoid and furanoid rings, respectively. (Hypophosphorous acid, one of the substitutes for tri-n-butyltin hydride mentioned in Appendix I, is the hydrogen-atom donor in the reaction shown in eq 6.) Xanthates also are used in Barton-McCombie reaction of alditols,29,30 cyclitols,31,32 and nucleosides.33,34
.png?revision=1&size=bestfit&width=335&height=115)
.png?revision=1&size=bestfit&width=410&height=107)
2. Thionocarbamates
Among thionocarbamates the (thiocarbonyl)imidazolides (3, Figure 1) are easily the most frequently used substrates for the Barton-McCombie reaction. The range of types of compounds involved is broad and includes (thiocarbonyl)imidazolides formed from carbohydrates with hydroxyl groups at the 2-,35 3‑,36–38 4-,39–41 and 6-42 positions in compounds with pyranoid rings, and at the 2‑,43,44 3-,45,46 and 5‑47,48 positions in compounds with furanoid rings. There also are numerous reports of Barton-McCombie reactions of nucleosides with O‑imidazol-1-ylthiocarbonyl groups at C-2'‑49,50 and C-3'.51,52
{kind=link}
3. Thionocarbonates
a. Phenyl Thionocarbonates
Phenyl thionocarbonates are yet another O-thiocarbonyl, carbohydrate derivative that undergoes the Barton-McCombie reaction. These derivatives participate in reaction at C-2,53,54 C-3,55,56 C-4,57,58 and C‑659,60 in compounds with pyranoid rings, and C-1,61,62 C-2,63,64 C‑3,65,66 and C‑567,68 in compounds with furanoid rings. Further, phenyl thionocarbonates are the preferred intermediates for nucleoside deoxygenation. They are involved in reaction at the 2'-position not only for a large number of 1,1,3,3-tetraisopropyl-1,3-disiloxanediyl-protected nucleosides,69,70 but also for compounds protected by benzyl,71 benzoyl,72,73t-butyldimethylsilyl,74 and pivaloyl groups.75 Similar reactions take place at the 3'- and 5'-positions.65,76
b. Substituted Phenyl Thionocarbonates
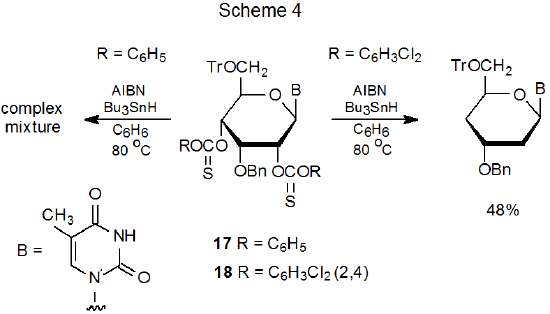
Phenyl thionocarbonates typically undergo a Barton-McCombie reaction that produces deoxygenated products in good yield; however, when product yields are low, introducing one or more electron-withdrawing substituents into the aromatic ring often raises these yields.77–80 Two examples illustrate the effect of these substituents. First, attempted Barton-McCombie reaction of the phenyl thionocarbonate 17 produces a complex mixture of products, but the 2,4-dichlorophenyl analog 18 forms the desired dideoxy nucleoside (Scheme 4).77 In the second example, the phenyl thionocarbonate 19 is unreactive under typical Barton-McCombie conditions, but its pentafluorophenyl analog 20 reacts with tri-n-butyltin hydride to give the corresponding deoxy nucleoside 21 (eq 8).78
.png?revision=1&size=bestfit&width=370&height=215)
In light of the better yields produced by the substituted phenyl thionocarbonates 18 and 20, it is surprising to find that the p-fluoro-, pentafluoro-, p-chloro-, 2,4,6-trichloro-, and pentachlorophenoxythiocarbonyl derivatives of cyclododecanol all react more slowly than the unsubstituted compound.6 In attempting to understand such a finding it is useful to consider the various ways in which ring substituents might influence reactivity. Evidence from the study of O-thiocarbonyl compounds suggests that the formation of the radical 23 (eq 9) is a reversible process and that the rate-determining step in this reaction sequence (equations 9 and 10) is the fragmentation of 23 shown in eq 10.9 An aromatic ring substituent is likely to impact this process in several ways. These include altering the radicophilicity of 22, the stability of 23, and the strength of the carbon–oxygen bond being broken to produce R· and 24 (eq 10); therefore, since an aromatic-ring substituent can exert influence on reactivity in several ways, understanding and predicting the overall effect that such a substituent will have on a reaction can be difficult.
.png?revision=1&size=bestfit&width=325&height=177)
c. Cyclic Thionocarbonates
Cyclic thionocarbonates undergoing Barton-McCombie reaction include compounds in which 2,3-O-thiocarbonyl,81–83 3,4-O-thiocarbonyl,53,84 and 4,6-O-thiocarbonyl85 groups are attached to pyranoid rings. This reaction also takes place with 2,3-O-thiocarbonyl7,8 and 5,6-O- thiocarbonyl67,86 groups in compounds with furanoid rings. Cyclic thionocarbonates, formed from acyclic structures, also undergo the Barton-McCombie reaction.87
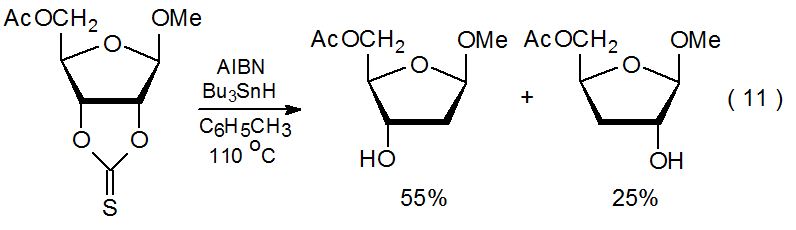
Reaction of a cyclic thionocarbonate is more complex than reaction of other O-thiocarbonyl compounds because it also involves ring opening. Since ring opening potentially can place a radical center on either of two carbon atoms, reaction often produces a mixture of products (eq 11).7,8 Formation of this mixture not only can reduce the amount of the desired product but also can complicate its isolation.
.png?revision=1&size=bestfit&width=400&height=116)
A proposed mechanism for reaction of a cyclic thionocarbonate with tri-n-butyltin hydride is given in Scheme 5.8 A "key" intermediate in this reaction is the radical 25, formed by addition of the tri-n-butyltin radical to the thiocarbonyl group. Radical stability usually controls the direction of ring opening; thus, even though 25 can produce either a primary or a secondary radical by ring opening, the pathway followed leads exclusively to the secondary radical (Scheme 5).7,8,88,89
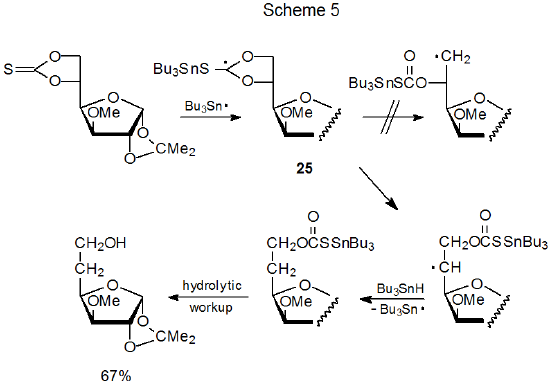
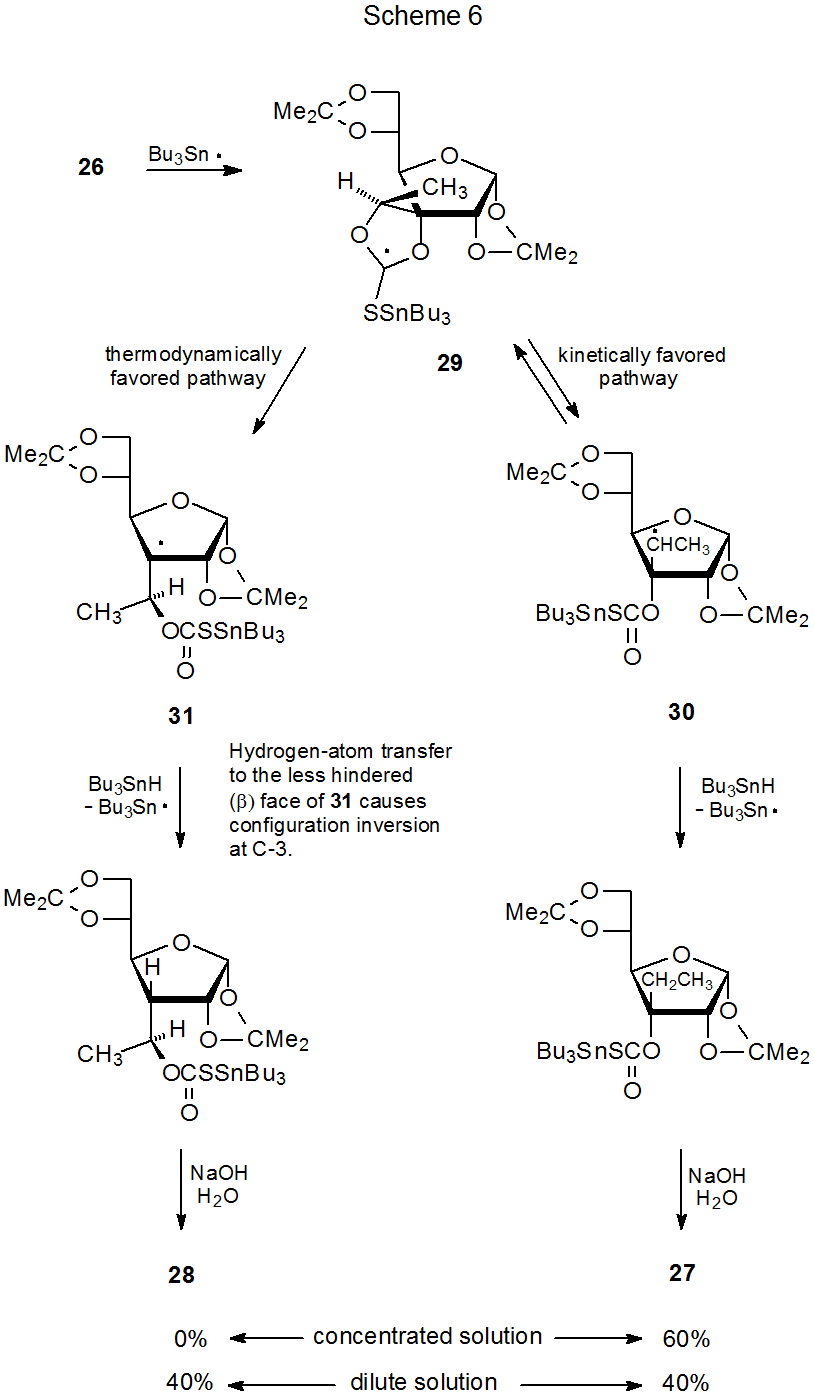
Although radical stability normally controls the direction of ring opening in a cyclic thionocarbonate, relief of angle strain in the transition state sometimes is the major factor.90 Ring opening of compound 26, for example, leads to the product derived from a secondary, rather than a tertiary, radical (eq 12).91 (A mechanism for this reaction is shown in Scheme 6.) Molecular mechanics calculations on noncarbohydrates indicate that fragmentation to give a less stable radical will occur if relief of ring strain in the transition state is great enough (eq 13).90 This relief of strain provides an explanation for the unexpected conversion of 26 into 27 rather than 28 (eq 12).
.png?revision=1&size=bestfit&width=430&height=202)
.png?revision=1&size=bestfit&width=425&height=159)
There is a concentration effect associated with the reaction of the cyclic thionocarbonate 26. In concentrated Bu3SnH solution only the kinetically favored product 27 is formed, but in dilute solution some of the thermodynamically favored isomer 28 is produced (Scheme 6). One explanation for this behavior is based on reversible formation of the secondary radical 30 from the cyclic radical 29. In concentrated solution 30 abstracts a hydrogen atom rapidly enough from Bu3SnH to prevent significant return to 29. Under these conditions only the product 27 is formed. In dilute solution hydrogen-atom abstraction by 30 is slowed to the point that reversible formation of 29 becomes significant and creates greater opportunity for 29 to be converted (irreversibly) into the thermodynamically favored tertiary radical 31. Since in dilute Bu3SnH solution both kinetically and thermodynamically favored pathways are followed, a mixture of the products 27 and 28 is produced.
4. Thionoesters
Primarily because their synthesis is more challenging, thionoesters are selected less frequently as starting materials for the Barton-McCombie reaction than are other O-thiocarbonyl derivatives. Thionoesters with O-thiocarbonyl groups at C-292 and C-31 in pyranoid rings and C-228 in furanoid rings are known substrates for deoxy sugar formation. A typical example is shown in eq 14.1 The thionoesters that undergo reaction are, with rare exception,93 thionobenzoates.28,92,94–101
.png?revision=1&size=bestfit&width=395&height=118)
E. Competing Reactions
Frequent application of the Barton-McCombie reaction in carbohydrate chemistry occurs because this reaction has a variety of attractive features. These include the broad range of compounds that undergo this reaction, the generally good product yields, and the freedom, in most cases, from significant, competing reactions. Even though side reactions usually do not represent a major concern, Barton-McCombie reaction has been conducted on so many compounds that quite a number of these reactions have been identified. They range in importance from alcohol regeneration, a common but usually minor side reaction, to phenyl-group migration, a rare event.
1. Alcohol Regeneration
Regenerating the alcohol from which an O-thiocarbonyl compound originally was synthesized is a side reaction sometimes accompanying deoxygenation. An example of such a reaction is shown in eq 15.102 In rare instances alcohol regeneration is the major reaction pathway (eq 16).42
.png?revision=1&size=bestfit&width=385&height=135)
.png?revision=1&size=bestfit&width=400&height=160)
Not all O-thiocarbonyl derivatives of carbohydrates are equally prone to alcohol regeneration. One of the advantages initially associated with phenyl thionocarbonates was that they did not undergo this reaction.3,4 Continued study of these compounds, however, showed that they are not immune to the alcohol-reforming process (eq 17).103
.png?revision=1&size=bestfit&width=435&height=173)
a. Proposed Reaction Mechanisms
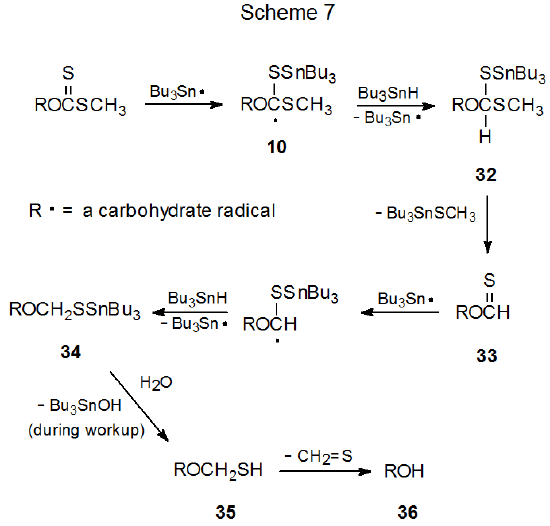
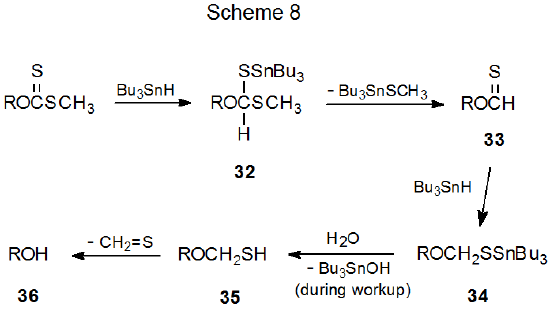
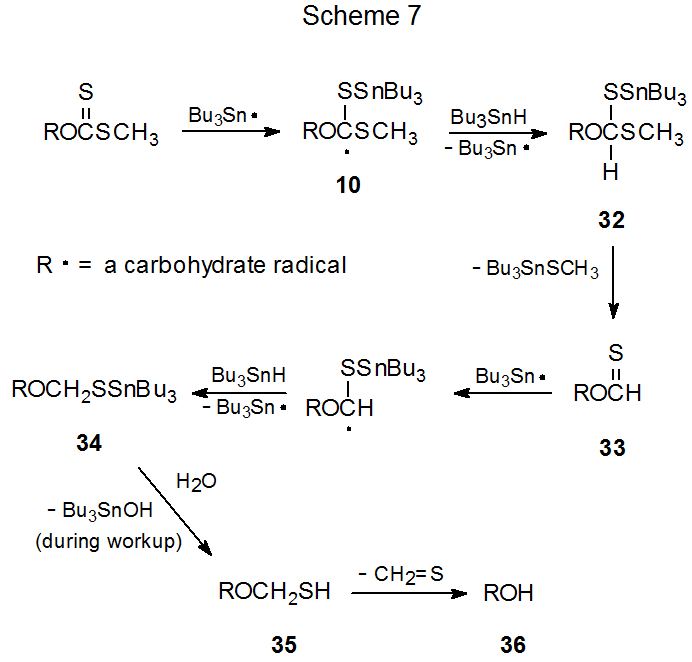
Although alcohol regeneration is the most common competing process during Barton-McCombie reaction, there is not general agreement on how the regeneration process proceeds. The two most frequently cited possibilities are both multi-step processes with many intermediates in common. The difference between these two rests with thiocarbonyl group reduction, one mechanism (Scheme 7) involves radical intermediates and the other (Scheme 8) does not.
(1). A Radical-Based Process
If one assumes that the radical 10 is an intermediate in the Barton-McCombie reaction (Scheme 2, X = SCH3), the possibility exists that before this radical fragments, it could abstract a hydrogen atom. Such a reaction would produce the intermediate 32 (Scheme 7).1,9 Formation of 32 not only would reduce the deoxygenated product yield, but it also could provide an explanation for the formation of the regenerated alcohol 36 as a side-product in the reaction.1,9 When viewed in this way, 32 is the beginning point in a series of events that leads first to the thionoformate 33, which reacts with tri-n-butyltin hydride to give the tin-containing intermediate 34, a substance that hydrolyzes during workup to the hemithioacetal 35. Compound 35 then decomposes spontaneously to give the alcohol 36 and thioformaldehyde (Scheme 7).9
{kind=link}
(2). A Hydride-Transfer-Based Reaction Sequence
The conditions originally used for the Barton-McCombie reaction did not include an added initiator; rather, reaction depended upon adventitious initiation. Within a few years, however, adding 2,2'-azobis(isobutyronitrile) (AIBN) became standard procedure because dependable initiation was recognized as a significant factor in maximizing deoxygenation.4,13 The reaction shown in eq 18 is one that provides an illustration of the improvement in product yield brought about by an added initiator.104 The observation that alcohol regeneration occurs in the absence of an initiator in the reaction shown in eq 18, supports the idea that a radical reaction may not be involved in the alcohol-reforming process.4
.png?revision=1&size=bestfit&width=420&height=154)
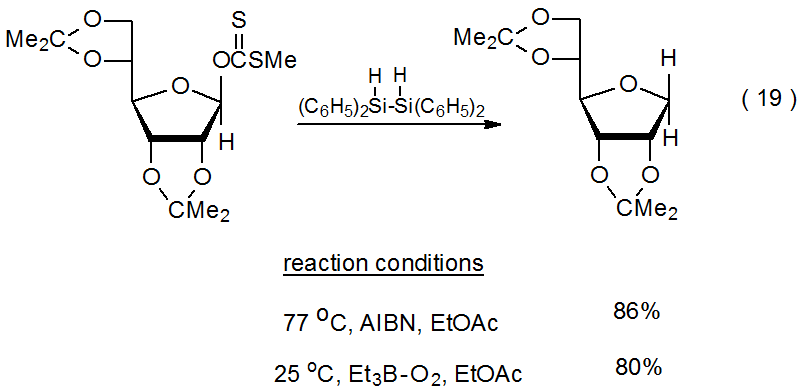
When the Barton-McCombie reaction is initiated by Et3B–O2,105,106 it can take place at temperatures much lower than those required for AIBN initiation (eq 19).21 Because it was originally thought that reducing the temperature of a Barton-McCombie reaction to about 80 oC caused alcohol regeneration to begin to become important,12 further lowering the reaction temperature would be expected to increase alcohol formation. When reactions were conducted at lower temperature using Et3B–O2 initiation, Barton-McCombie reaction took place with little or no alcohol regeneration. This finding was not consistent with the idea that 10 (Scheme 7) was increasingly likely to abstract a hydrogen atom from Bu3SnH as the reaction temperature decreased.13 If the conversion of 10 into 32 by hydrogen-atom abstraction were not taking place, it raised the possibility that 32 was formed in a different, perhaps nonradical, reaction (Scheme 8).
{kind=link}
.png?revision=1&size=bestfit&width=405&height=195)
Several investigators have proposed that hydride transfer may be responsible for alcohol regeneration.13,107,108 As a part of the most detailed of these proposals, it was suggested that nonradical addition of tri-n-butyltin hydride to an O‑thiocarbonyl group occurs in the first step in alcohol regeneration, and it occurs later in the reaction sequence when the thionoformate 33 is converted into the thioacetal 34 (Scheme 8).108
b. Alcohols from Dimeric Esters
The observation that minor amounts of “dimeric’ esters were formed during the phenyl thionocarbonate synthesis shown in eq 20 raised the possibility that if these esters were not removed prior to Barton-McCombie reaction, an alcohol could be formed because each dimeric ester reasonably could be expected to react with tri-n-butyltin hydride to give a molecule of a deoxygenated compound and one of the starting alcohol.103 This expectation was confirmed when the dimeric ester 37 was found to undergo the reaction shown in eq 21.109
.png?revision=1&size=bestfit&width=405&height=159)
.png?revision=1&size=bestfit&width=280&height=297)
c. Minimizing Alcohol Regeneration
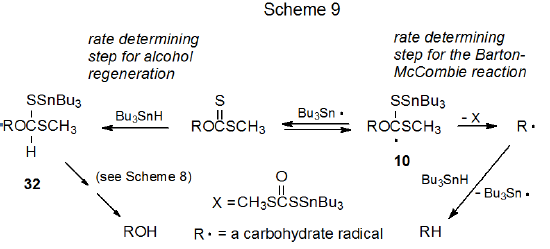
The rate determining step in the Barton-McCombie reaction is believed to be the unimolecular fragmentation of the radical 10 to give the carbon-centered radical R· (Scheme 9).9 For alcohol regeneration, however, the rate is more likely to depend upon a bimolecular reaction involving Bu3SnH. This analysis (Scheme 9) is consistent with the finding that keeping the concentration of Bu3SnH at a low level during reaction (i.e., adding the tin hydride slowly as the reaction proceeds) is associated with maximizing deoxygenation; for example, the xanthate shown in eq 22 reacts to give a deoxy sugar in 54% yield when all the Bu3SnH is present at the beginning of the reaction, but the yield rises to 85% when Bu3SnH is added over a period of 1.5 h.36 Part of the reduced deoxy sugar yield in the reaction where all the Bu3SnH was added at the beginning is due to recovery of the alcohol from which the xanthate was synthesized.36
.png?revision=1&size=bestfit&width=355&height=181)
2. O-Thionocarbonyl Group Conversions and Rearrangements
a. Conversion of a Xanthate into a Dithiocarbonate
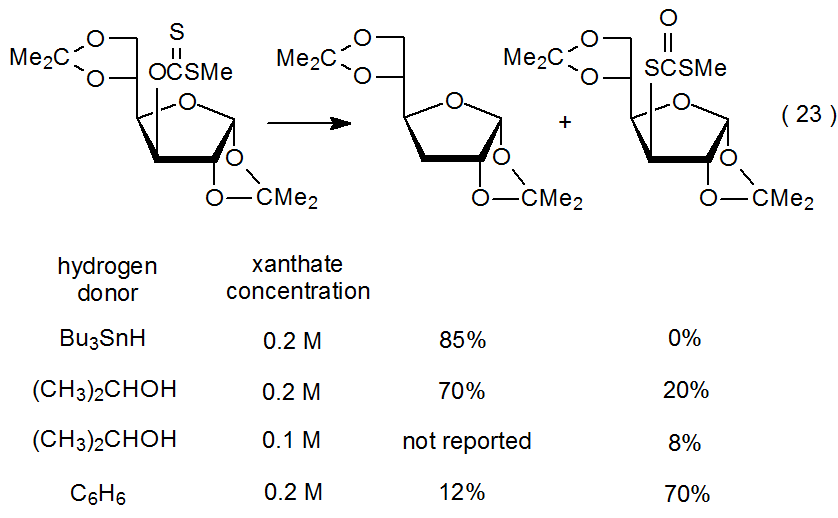
When Bu3SnH is the hydrogen-atom donor in the reaction shown in eq 23,1 a deoxy sugar forms in the normal manner, but if a less effective donor is used, xanthate conversion to a dithiocarbonate competes with the deoxygenation process. This conversion becomes significant when reaction is conducted with 2‑propanol serving as both solvent and hydrogen-atom donor. When benzene is the solvent, dithiocarbonate formation is the major reaction pathway.110 Benzene is, in fact, such a poor hydrogen-atom donor that any carbohydrate present in solution is a more likely hydrogen-atom source for the small amount of deoxy sugar formed.
.png?revision=1&size=bestfit&width=420&height=260)
The reaction shown in eq 23 begins with formation of the carbohydrate radical R·. As pictured in Scheme 10, this radical (R·) then either adds to a molecule of starting material, leading to a dithiocarbonate, or abstracts a hydrogen atom, producing a deoxy sugar. The data presented in eq 23 confirm the expectation from the proposed mechanism (Scheme 10) that deoxy sugar formation is favored when effective hydrogen-atom donors are used, and dithiocarbonates form more easily in reactions run at high xanthate concentrations in the presence of poor hydrogen-atom donors.110
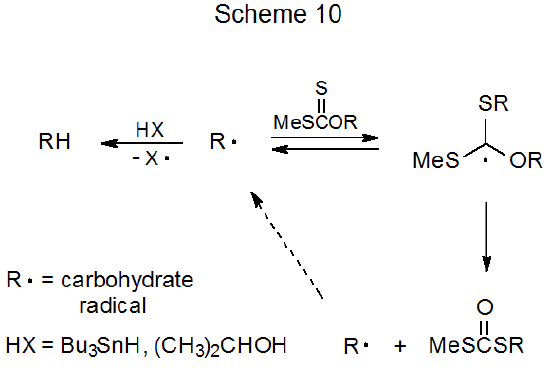
b. Thionocarbonate-Thiocarbonate Rearrangement
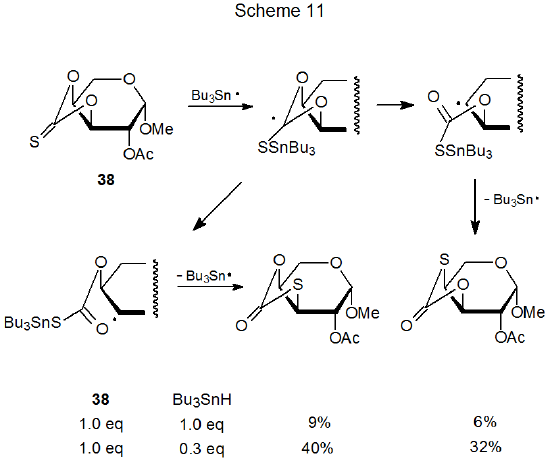
A reaction closely related to the xanthate-dithiocarbonate rearrangement just discussed is the conversion of a thionocarbonate into a thiocarbonate. When a hydrogen-atom donor is present in a reaction mixture in an amount less than that needed to supply a hydrogen atom to each carbohydrate radical, thionocarbonate to thiocarbonate rearrangement can take place.82,88,89,111,112 In the reaction shown in Scheme 11 this rearrangement represents the major reaction pathway when the amount of tri-n-butyltin hydride is significantly less than that required for complete reduction.111 Rearrangement is, of course, undesirable when simple reduction is the goal of a reaction, but it can be useful if the purpose of the reaction is to convert a sugar into a thiosugar.112
c. Conversion of an S-Alkyl Xanthate into an O-Alkyl Xanthate
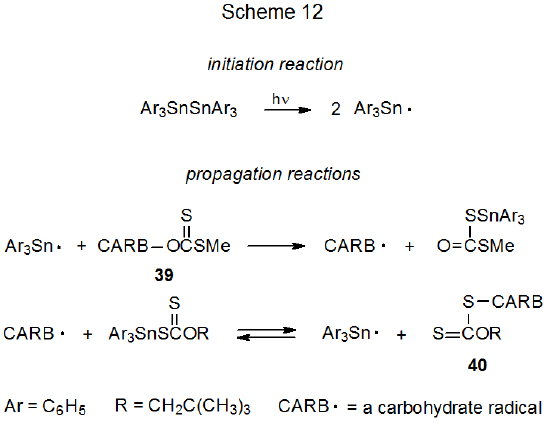
Reaction of the carbohydrate xanthate 39 (an S-alkyl xanthate) with the triphenyltin radical is the first step in a propagation sequence (Scheme 12) that converts 39 into a xanthate with the carbohydrate portion of the molecule bonded to sulfur (40, an O-alkyl xanthate).113,114 The second step in this sequence is reaction of the carbohydrate radical with triphenyltin xanthate to produce 40 and the chain-carrying, triphenyltin radical. This second step must be reversible in order to account for the epimers 41 and 42 being interconverted under the reaction conditions (eq 24).114 Effective hydrogen-atom donors, such as triphenyltin hydride, must be excluded to prevent simple reduction. Excluding triphenyltin hydride but still having the triphenyltin radical needed to initiate the reaction is accomplished by photolysis of bis(triphenyltin) (Scheme 12).
.png?revision=1&size=bestfit&width=425&height=159)
3. Reaction With Molecular Oxygen
Reaction conducted in the presence of molecular oxygen leads to rapid capture of O2 by carbon-centered radicals. This capture is faster than hydrogen-atom abstraction from Bu3SnH. Radical capture of O2 is suppressed by the normal procedure of excluding oxygen from the reaction mixture, but when O2 is deliberately added, its combination with a carbohydrate radical becomes a major or even the exclusive reaction pathway (eq 25).89
.png?revision=1&size=bestfit&width=400&height=179)
4. Elimination Reactions
a. Reactions of Compounds with Two O-Thiocarbonyl Groups
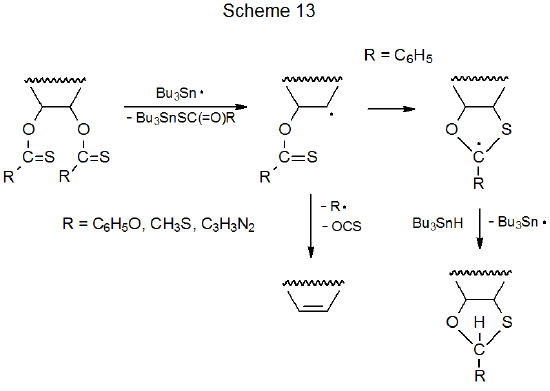
If there are two O-thiocarbonyl groups in the same molecule, their physical separation affects whether or not Barton-McCombie reaction will take place.1,115–134 In the reaction of compound 43, for example, the substituents are sufficiently well separated to allow replacement of each group by a hydrogen atom to proceed in the normal manner (eq 26115).115,130 When O‑thiocarbonyl groups are attached to adjacent carbon atoms, reaction of one of these groups produces a carbon-centered radical that forms a double bond by elimination of the second group.117–129,131,133 An example is shown in eq 27.117–119 As indicated in Scheme 13, elimination takes place when R = OC6H5 or SCH3, but in the rare event that R = C6H5, the elimination pathway leads to the unstable phenyl radical; as a consequence, radical cyclization takes place instead of elimination (eq 28).1 If two O-thiocarbonyl groups are not on adjacent carbon atoms, but the radical produced by reaction of the first is centered on an atom in close proximity to the second, internal addition will take place.115,116,132,134 Because the new radical formed by this reaction does not have a clear path to an elimination product, more complex reaction, such as that shown in eq 29,115 is likely to take place.
.png?revision=1&size=bestfit&width=410&height=114)
.png?revision=1&size=bestfit&width=380&height=136)
.png?revision=1&size=bestfit&width=420&height=122)
.png?revision=1&size=bestfit&width=405&height=128)
b. Reactions of Compounds with a Single O-Thiocarbonyl Group
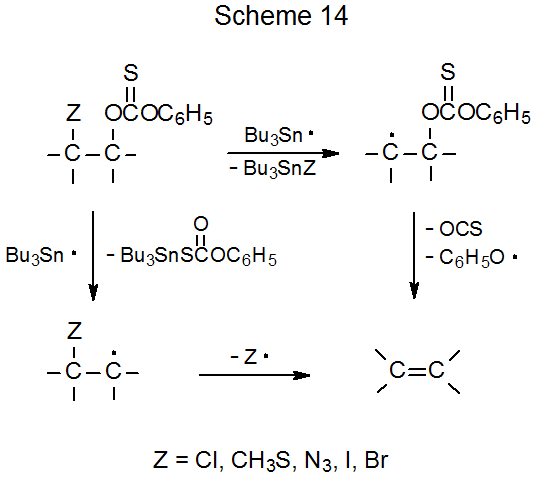
As described in the previous section, elimination reactions take place when compounds with O-thiocarbonyl groups on adjacent carbon atoms react with tin or silicon hydrides. Similar reaction takes place when an O-thiocarbonyl group is attached to a carbon atom that has an azido,135 bromo,135–137 chloro,135,138,139 iodo,135 isocyano,140 methylthio,135 or phenylthio,141 substituent bonded to an adjacent carbon atom. An example is given in eq 30.135 This process begins with formation of a carbon-centered radical and ends with radical expulsion from an adjacent carbon atom (Scheme 14). In some instances it is reaction of the O‑thiocarbonyl group that generates the carbon-centered radical, but in others, particularly those involving compounds containing bromine and iodine, halogen-atom abstraction is the first reaction to take place.
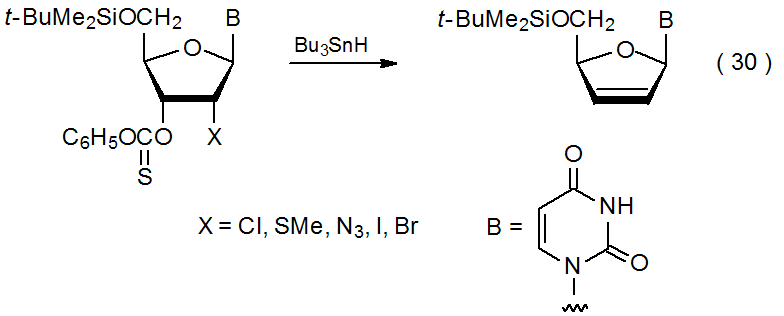.png?revision=1&size=bestfit&width=395&height=159)
c. Use of Sacrificial Olefins
One of the difficulties associated with the synthesis of unsaturated compounds by radical reaction is that a radical intermediate may add to a recently formed double bond in a product molecule. When such an unwanted addition occurs, it may be reduced in importance to an acceptable level by adding an alkene such as 1-dodecene to the reaction mixture. This alkene, a compound described as a “sacrificial olefin”, protects the elimination product by scavenging radicals before they add to product molecules.142
d. Cyclic Thionocarbonates
One way to view cyclic thionocarbonates is as compounds that have adjacent O-thiocarbonyl groups and, consequently, might undergo radical elimination. Since Barton-McCombie reactions of cyclic thionocarbonates generally give good yields of deoxy compounds, an elimination reaction can be expected only if such a reaction benefits from a special driving force. This driving force can come from reaction producing an unsaturated compound with a double bond stable enough that its formation significantly lowers transition-state energy. Consistent with this idea is formation of the glycal 46 as the only product from reaction of the cyclic thionocarbonate 45 with tri-n-butyltin hydride (eq 31).143 2',3'-O-Thiocarbonyl nucleoside derivatives undergo similar reaction to give unsaturated nucleosides, but only as minor products.144–146
.png?revision=1&size=bestfit&width=425&height=121)
e. Preventing Thermal Elimination
The Barton-McCombie reaction of xanthates is most successful when these compounds are prepared from secondary alcohols. Xanthates formed from tertiary alcohols are prone to thermal elimination (eq 32, Chugaev elimination147) at temperatures normally used in the Barton-McCombie reaction. When reaction is initiated by triethylboron–oxygen, however, it can be conducted at room temperature, where reaction of tertiary xanthates occurs without competing elimination.106,148
.png?revision=1&size=bestfit&width=405&height=85)
5. Reversible Addition to an O-Thiocarbonyl Group
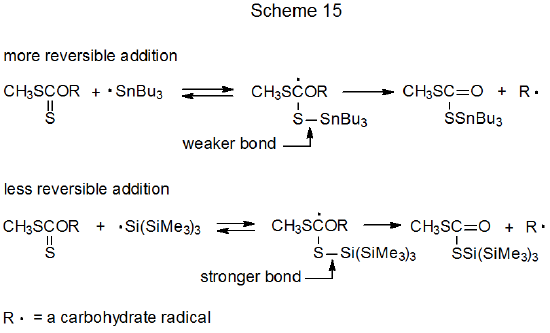
Xanthates synthesized from primary alcohols usually require higher temperatures in the Barton-McCombie reaction and often give lower product yields.149,150 These problems can be overcome in some instances by changing the hydrogen-atom donor from Bu3SnH to (Me3Si)3SiH. Tri-n-butyltin hydride is a less effective donor than tris(trimethylsilyl)silane in these reactions due to greater reversibility of Bu3Sn· addition to a thiocarbonyl group (Scheme 15).149 Because the S–Si bond [90 kcal mol-1 (377 kJ mol-1)]151 is stronger than the S–Sn bond [65 kcal mol-1 (272 kJ mol‑1)],151 reversal of addition of (Me3Si)3Si· is less likely to occur. Reduced reversibility means that once a silyl radical has added to an O-thiocarbonyl group, a difficult forward reaction (e.g., one producing a primary radical149 and, sometimes, one producing a secondary radical152,153 ) can compete more effectively with reverse reaction to give the starting materials. Limiting reversibility is even more effective when reaction take place under the low temperature conditions made possible by Et3B–O2 initiation.33
6. Reduction of a Thiocarbonyl Group to a Methylene Group
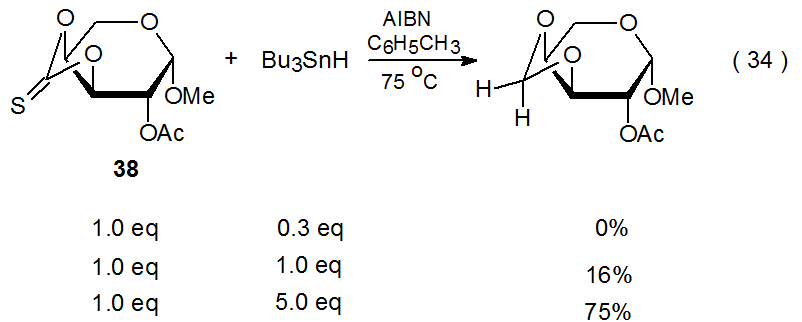
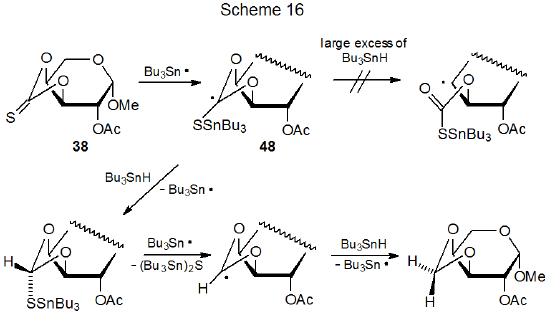
Conversion of a thiocarbonyl group into a methylene group represents a rare type of competition for the Barton-McCombie reaction. This transformation changes the O‑phenylthiocarbonyl group in 47 into an O-benzyl group (eq 33),28 and it is responsible for a similar change in the cyclic thionocarbonate 38 (eq 34).89,111 The reaction shown in eq 34 occurs in much higher yield when a large excess of tri-n-butyltin hydride is present. One effect of a large excess of Bu3SnH is to increase the rate of hydrogen-atom abstraction by the radical 48 (Scheme 16) to the point that ring opening by this radical is too slow to be detected.
.png?revision=1&size=bestfit&width=335&height=120)
.png?revision=1&size=bestfit&width=405&height=163)
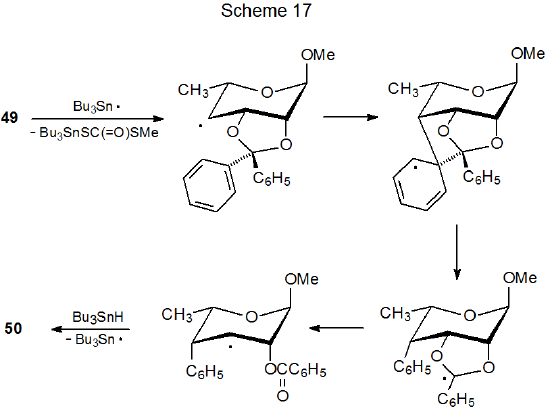
7. Phenyl-Group Migration
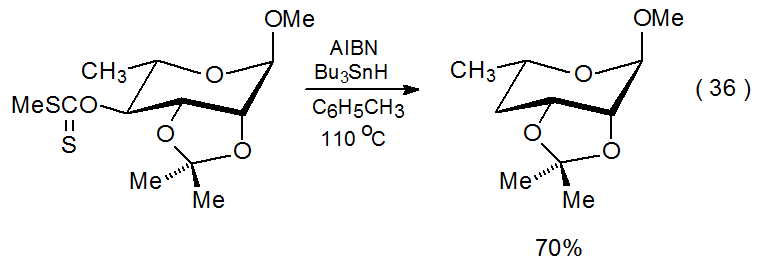
Compound 49, a xanthate with an O-[(alkylthio)thiocarbonyl] group at C-4 and 2,3-O-diphenylmethylene protection, undergoes phenyl group migration and ring opening during reaction (eq 35).154 According to the mechanism proposed in Scheme 17, this reaction depends upon the presence of substituents in the protecting group that can undergo migration by an addition-elimination process. Consistent with this mechanistic proposal is the observation that if the 2,3-O-diphenylmethylene group is replaced by a 2,3-O-isopropylidene group, normal reaction occurs (eq 36).155
.png?revision=1&size=bestfit&width=430&height=133)
.png?revision=1&size=bestfit&width=385&height=134)
F. Influence of Steric Effects on Reactivity
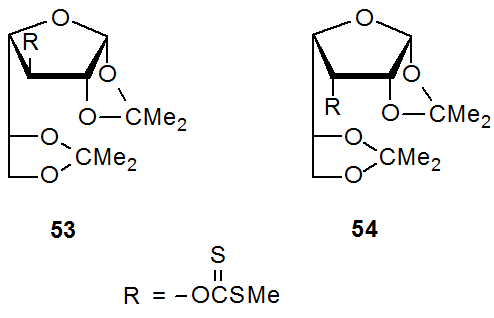
Reaction of the epimeric thionocarbamates 51 and 52 with Bu3SnH illustrates the role that steric effects play in the Barton-McCombie reaction (eq 37).156 The dramatically different steric environment for the C-3 substituents in 51 and 52 does not have a substantial impact on their reactivity; specifically, the difference of a factor of four in reaction times (reaction rates were not measured) is consistent with only minor influence by steric effects. A possible explanation for this modest difference in reactivity is that although the steric congestion around C-3 in compounds 51 and 52 is substantially different, it is not dramatically different in the area of the sulfur atom, where the reaction is taking place. Comparing the reactivity of the xanthates 53 and 54 provides another example of the modest role of steric effects in the Barton-McCombie reaction. The decidedly more hindered O-[(methylthio)thiocarbonyl] group in 54 renders this xanthate (54) only about five times less reactive than its epimer 53.157
.png?revision=1&size=bestfit&width=425&height=194)
G. Regioselectivity
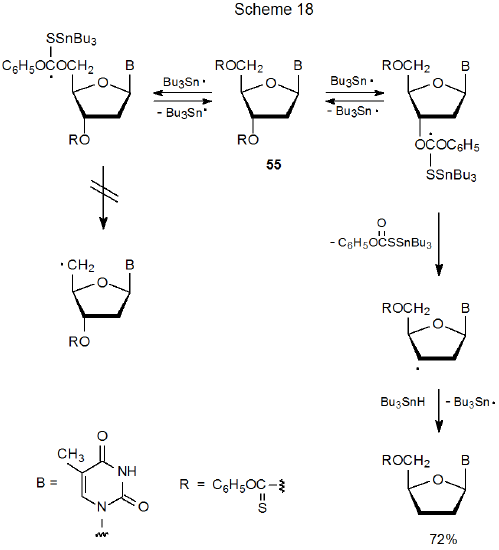
One of the characteristics of the Barton-McCombie reaction is that when it is conducted in the normal manner (i.e., with Bu3SnH as the hydrogen-atom donor and AIBN as the initiator), higher temperature is required for reaction of a primary O-phenoxythiocarbonyl group than a secondary one.76,158 This difference in reactivity can become the basis for regioselective reaction; thus, as pictured in Scheme 18, group replacement takes place only at the 3'‑position in the nucleoside 55.76
H. Chemoselectivity
Chemoselectivity in reactions of compounds containing O-thiocarbonyl groups is discussed in Section II.B of Chapter 9 in Volume I.
I. The β-Oxygen Effect
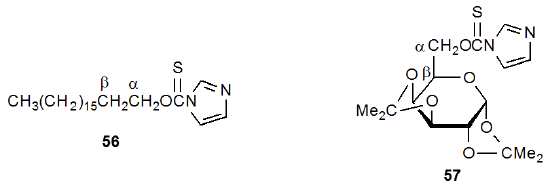
An observation about the Barton-McCombie reaction is that some carbohydrates react more easily than would be expected on the basis of model compound behavior. This greater reactivity is observed when the carbon atom in a C–O bond is β-related to a carbon atom bearing an O-thiocarbonyl group.93 Comparison of the reactivities of 56 and 57 illustrates this difference. Compound 56 does not react with tri-n-butyltin hydride at 110 oC (it does at 130 oC),93 but 57, which also is a primary (thiocarbonyl)imidazolide, does react under these conditions42,93 This and similar observations led to the proposal that "oxygen bonded to the β-carbon of a carbon radical has a marked stabilizing effect; this permits radical reactions not seen, except at much higher temperatures, in non-oxygenated model compounds."93 When first proposed, this "β-oxygen effect" represented a potentially important factor in carbohydrate chemistry because many carbon-centered radicals would be stabilized by its existence.
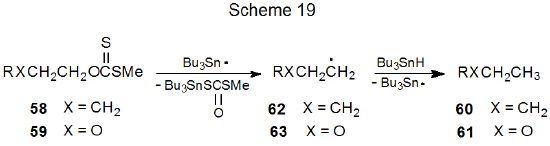
Considerable effort has been invested in studying the β-oxygen effect. This work has established that the presence of a β-related, carbon–oxygen bond does not necessarily increase the rate of formation of a developing, carbon-centered radical. For example, reaction of a mixture of the xanthates 58 and 59 with a limited amount of tri-n-butyltin hydride gave products 60 and 61 in approximately equal amounts (Scheme 19).159,160 Since this result meant that xanthates 58 and 59 were reacting at essentially the same rate, a β-related, carbon–oxygen bond was providing little, if any, transition-state stabilization for the developing radical 63.160
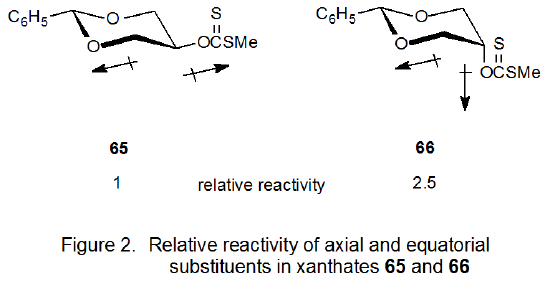
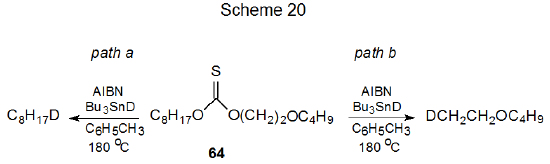
A related experiment involved the thionocarbonate 64, which could react along either of two competing pathways (Scheme 20). If the β-oxygen effect existed, reaction by path b would be favored. Experimentation showed that reaction along each pathway was equally likely; consequently, the conclusion again was that no evidence existed for a β-oxygen effect.149
Although these experiments dispelled the idea that a β-related oxygen atom generally provides stabilization to a radical center, they did not explain why the carbohydrate derivative 57 is more reactive than the model compound 56. An explanation for this behavior, however, does come from study of the equatorial and axial isomers 65 and 66, respectively. The axial epimer (66) with its gauche (synclinal) dipoles is more reactive than the equatorial epimer (65) with trans dipoles (Figure 2). The greater reactivity of compound 66 is attributed to eliminating an unfavorable dipole-dipole interaction during bond breaking. Removing such an interaction then appears to be the primary reason for the rate differences that originally were attributed to the β-oxygen effect.149