8.4: Electrophiles and carbocation stability
- Page ID
- 1085

In the vast majority of the nucleophilic substitution reactions you will see in this and other organic chemistry texts, the electrophilic atom is a carbon which is bonded to an electronegative atom, usually oxygen, nitrogen, sulfur, or a halogen. The concept of electrophilicity is relatively simple: an electron-poor atom is an attractive target for something that is electron-rich, i.e. a nucleophile. However, we must also consider the effect of steric hindrance on electrophilicity. In addition, we must discuss how the nature of the electrophilic carbon, and more specifically the stability of a potential carbocationic intermediate, influences the SN1 vs. SN2 character of a nucleophilic substitution reaction.
8.4A: Steric effects on electrophilicity
Consider two hypothetical SN2 reactions: one in which the electrophile is a methyl carbon and another in which it is tertiary carbon.
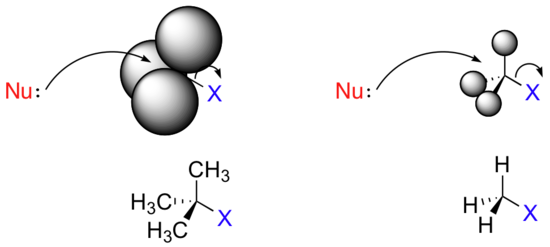
Because the three substituents on the methyl carbon electrophile are tiny hydrogens, the nucleophile has a relatively clear path for backside attack. However, backside attack on the tertiary carbon is blocked by the bulkier methyl groups. Once again, steric hindrance - this time caused by bulky groups attached to the electrophile rather than to the nucleophile - hinders the progress of an associative nucleophilic (SN2) displacement.
The factors discussed in the above paragraph, however, do not prevent a sterically-hindered carbon from being a good electrophile - they only make it less likely to be attacked in a concerted SN2 reaction. Nucleophilic substitution reactions in which the electrophilic carbon is sterically hindered are more likely to occur by a two-step, dissociative (SN1) mechanism. This makes perfect sense from a geometric point of view: the limitations imposed by sterics are significant mainly in an SN2 displacement, when the electrophile being attacked is a sp3-hybridized tetrahedral carbon with its relatively ‘tight’ angles of 109.4o. Remember that in an SN1 mechanism, the nucleophile attacks an sp2-hybridized carbocation intermediate, which has trigonal planar geometry with ‘open’ 120 angles.

With this open geometry, the empty p orbital of the electrophilic carbocation is no longer significantly shielded from the approaching nucleophile by the bulky alkyl groups. A carbocation is a very potent electrophile, and the nucleophilic step occurs very rapidly compared to the first (ionization) step.
8.4B: Stability of carbocation intermediates
We know that the rate-limiting step of an SN1 reaction is the first step - formation of the this carbocation intermediate. The rate of this step – and therefore, the rate of the overall substitution reaction – depends on the activation energy for the process in which the bond between the carbon and the leaving group breaks and a carbocation forms. According to Hammond’s postulate (section 6.2B), the more stable the carbocation intermediate is, the faster this first bond-breaking step will occur. In other words, the likelihood of a nucleophilic substitution reaction proceeding by a dissociative (SN1) mechanism depends to a large degree on the stability of the carbocation intermediate that forms.
The critical question now becomes, what stabilizes a carbocation?
Think back to Chapter 7, when we were learning how to evaluate the strength of an acid. The critical question was “how stable is the conjugate base that results when this acid donates its proton"? In many cases, this conjugate base was an anion – a center of excess electron density. Anything that can draw some of this electron density away– in other words, any electron withdrawing group – will stabilize the anion.
So if it takes an electron withdrawing group to stabilize a negative charge, what will stabilize a positive charge? An electron donating group!
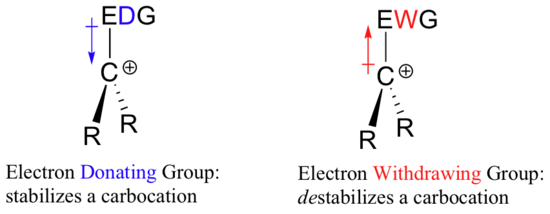
A positively charged species such as a carbocation is very electron-poor, and thus anything which donates electron density to the center of electron poverty will help to stabilize it. Conversely, a carbocation will be destabilized by an electron withdrawing group.
Alkyl groups – methyl, ethyl, and the like – are weak electron donating groups, and thus stabilize nearby carbocations. What this means is that, in general, more substituted carbocations are more stable: a tert-butyl carbocation, for example, is more stable than an isopropyl carbocation. Primary carbocations are highly unstable and not often observed as reaction intermediates; methyl carbocations are even less stable.
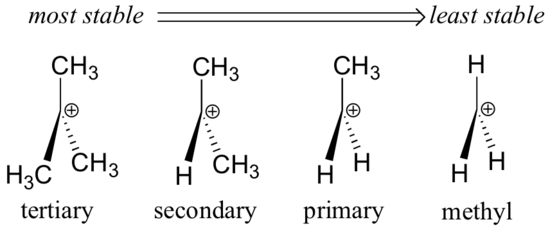
Alkyl groups are electron donating and carbocation-stabilizing because the electrons around the neighboring carbons are drawn towards the nearby positive charge, thus slightly reducing the electron poverty of the positively-charged carbon.
It is not accurate to say, however, that carbocations with higher substitution are always more stable than those with less substitution. Just as electron-donating groups can stabilize a carbocation, electron-withdrawing groups act to destabilize carbocations. Carbonyl groups are electron-withdrawing by inductive effects, due to the polarity of the C=O double bond. It is possible to demonstrate in the laboratory (see section 16.1D) that carbocation A below is more stable than carbocation B, even though A is a primary carbocation and B is secondary.
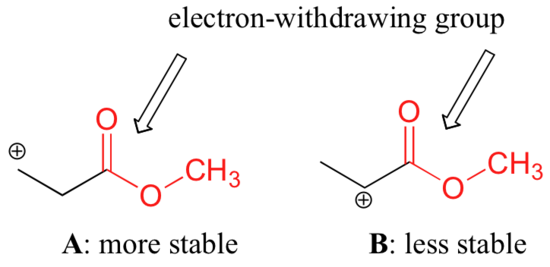
The difference in stability can be explained by considering the electron-withdrawing inductive effect of the ester carbonyl. Recall that inductive effects - whether electron-withdrawing or donating - are relayed through covalent bonds and that the strength of the effect decreases rapidly as the number of intermediary bonds increases. In other words, the effect decreases with distance. In species B the positive charge is closer to the carbonyl group, thus the destabilizing electron-withdrawing effect is stronger than it is in species A.
In the next chapter we will see how the carbocation-destabilizing effect of electron-withdrawing fluorine substituents can be used in experiments designed to address the question of whether a biochemical nucleophilic substitution reaction is SN1 or SN2.
Stabilization of a carbocation can also occur through resonance effects, and as we have already discussed in the acid-base chapter, resonance effects as a rule are more powerful than inductive effects. Consider the simple case of a benzylic carbocation:
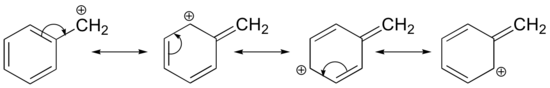
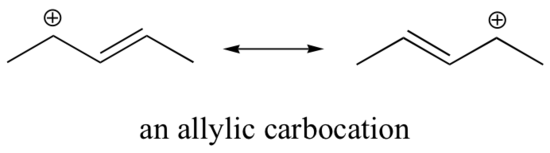
This carbocation is comparatively stable. In this case, electron donation is a resonance effect. Three additional resonance structures can be drawn for this carbocation in which the positive charge is located on one of three aromatic carbons. The positive charge is not isolated on the benzylic carbon, rather it is delocalized around the aromatic structure: this delocalization of charge results in significant stabilization. As a result, benzylic and allylic carbocations (where the positively charged carbon is conjugated to one or more non-aromatic double bonds) are significantly more stable than even tertiary alkyl carbocations.
Because heteroatoms such as oxygen and nitrogen are more electronegative than carbon, you might expect that they would by definition be electron withdrawing groups that destabilize carbocations. In fact, the opposite is often true: if the oxygen or nitrogen atom is in the correct position, the overall effect is carbocation stabilization. This is due to the fact that although these heteroatoms are electron withdrawing groups by induction, they are electron donating groups by resonance, and it is this resonance effect which is more powerful. (We previously encountered this same idea when considering the relative acidity and basicity of phenols and aromatic amines in section 7.4). Consider the two pairs of carbocation species below:
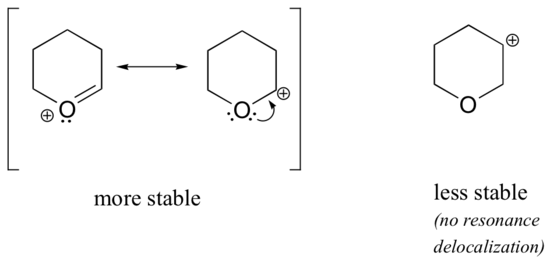
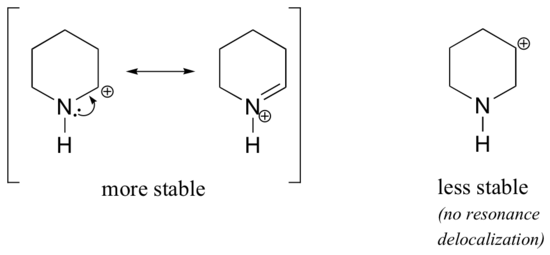
In the more stable carbocations, the heteroatom acts as an electron donating group by resonance: in effect, the lone pair on the heteroatom is available to delocalize the positive charge. In the less stable carbocations the positively-charged carbon is more than one bond away from the heteroatom, and thus no resonance effects are possible. In fact, in these carbocation species the heteroatoms actually destabilize the positive charge, because they are electron withdrawing by induction.
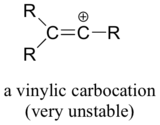
Finally, vinylic carbocations, in which the positive charge resides on a double-bonded carbon, are very unstable and thus unlikely to form as intermediates in any reaction.
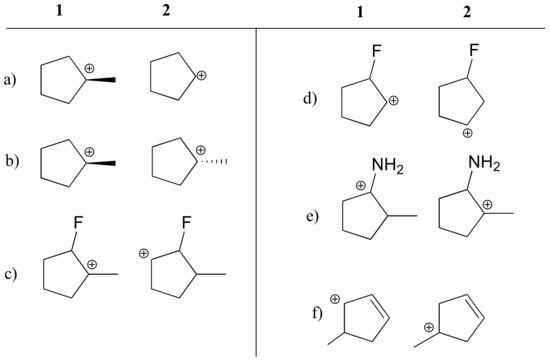
| Example 8.10 |
|---|
| Exercise 8.10: In which of the structures below is the carbocation expected to be more stable? Explain.
|
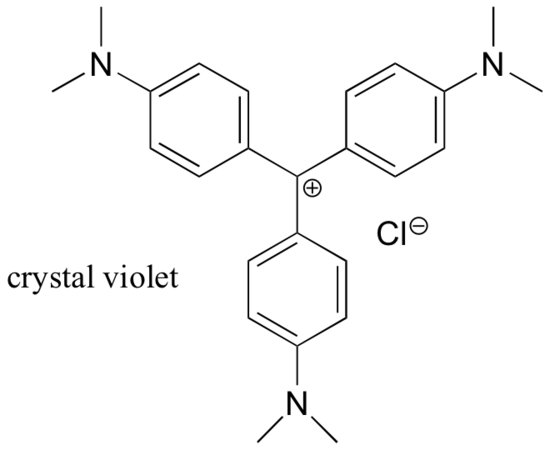
For the most part, carbocations are very high-energy, transient intermediate species in organic reactions. However, there are some unusual examples of very stable carbocations that take the form of organic salts. Crystal violet is the common name for the chloride salt of the carbocation whose structure is shown below. Notice the structural possibilities for extensive resonance delocalization of the positive charge, and the presence of three electron-donating amine groups.
| Example 8.11 |
|---|
| Draw a resonance structure of the crystal violet cation in which the positive charge is delocalized to one of the nitrogen atoms. |
When considering the possibility that a nucleophilic substitution reaction proceeds via an SN1 pathway, it is critical to evaluate the stability of the hypothetical carbocation intermediate. If this intermediate is not sufficiently stable, an SN1 mechanism must be considered unlikely, and the reaction probably proceeds by an SN2 mechanism. In the next chapter we will see several examples of biologically important SN1 reactions in which the positively charged intermediate is stabilized by inductive and resonance effects inherent in its own molecular structure.
| Example 8.12 |
|---|
| State which carbocation in each pair below is more stable, or if they are expected to be approximately equal. Explain your reasoning.
|