
10.1: Overview of phosphates and phosphoryl transfer reactions
- Page ID
- 953

10.1A: Nomenclature and abbreviations
Phosphoryl groups are derivatives of phosphoric acid, a strong acid that is commonly used in the laboratory. The fully deprotonated conjugate base of phosphoric acid is called a phosphate ion, or inorganic phosphate (often abbreviated 'Pi'). When two phosphate groups are linked to each other, the linkage is referred to as a 'phosphate anhydride', and the ion is called 'inorganic pyrophosphate' (abbreviation PPi).
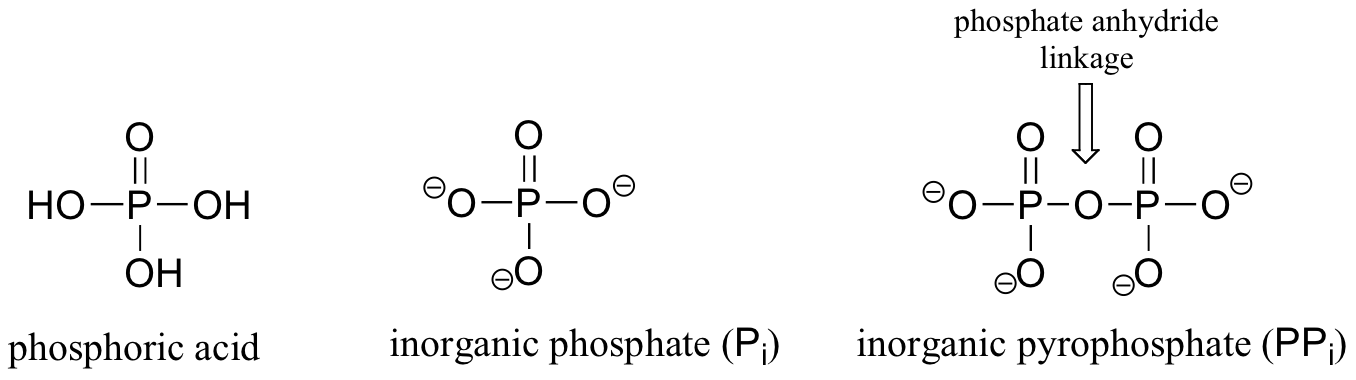
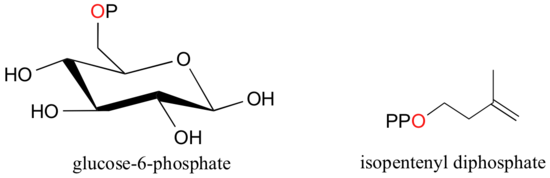
When a phosphate ion is attached to a carbon atom on an organic molecule, the chemical linkage is referred to as a phosphate ester, and the whole species is called an organic monophosphate. Glucose-6-phosphate is an example.

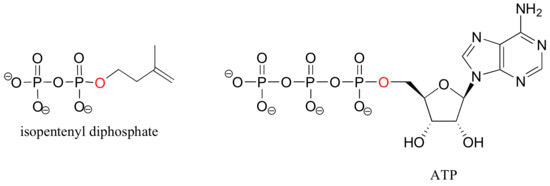
If an organic molecule is linked to two or three phosphate groups, the resulting species are called organic diphosphates and organic triphosphates.

Isopententyl diphosphate and adenosine triphosphate (ATP) are good examples:
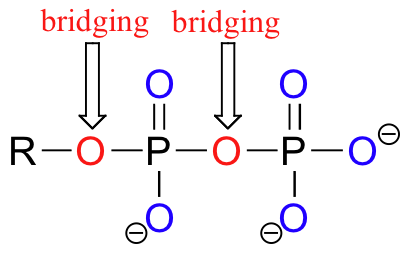
Oxygen atoms in phosphate groups are referred to either 'bridging' and 'non-bridging', depending on their position. An organic diphosphate has two bridging and five non-bridging oxygens.
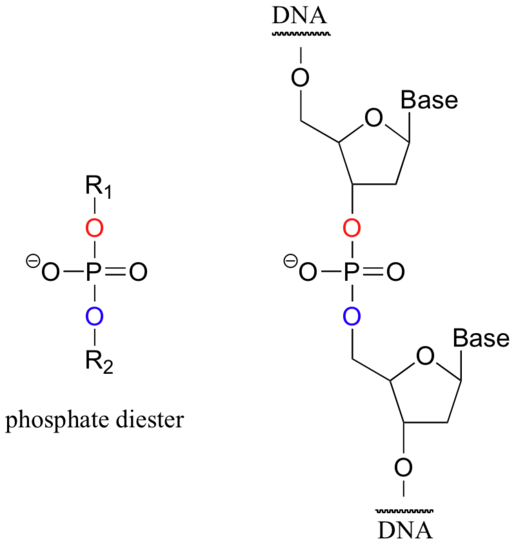
When a single phosphate is linked to two organic groups, the term 'phosphate diester' is used. The backbone of DNA is composed of phosphate diesters.
The term 'phosphoryl group' is a general way to refer to all of the phosphate-based groups mentioned in the paragraphs above.
Recall (section 1.4A) that phosphate groups on organic structures are sometimes abbreviated simply as 'P', a convention that we will use throughout this text. For example, glucose-6-phosphate and isopentenyl diphosphate are often depicted as shown below. Notice that the 'P' abbreviation includes the oxygen atoms and negative charges associated with the phosphate groups.
10.1B: Acid constants and protonation states
Phosphoric acid is triprotic, meaning that it has three acidic hydrogens available to donate, with pKa values of 2.1, 7.2, and 12.3, respectively. These acid constant values tell us that, at the physiological pH of approximately 7.3, all phosphoric acid species in solution will have donated at least one proton, and more than half will have donated two, meaning that the average charge on the phosphate ion is slightly higher than -1.5. Organic monophosphates, diphosphates, and triphosphates are predominantly deprotonated at pH 7.3, meaning that they carry charges of slightly less than -2, -3, and -4, respectively. By convention, they are usually drawn in their fully deprotonated states.
10.1C: Bonding in phosphines and phosphates
Looking at the location of phosphorus on the periodic table, you might expect it to bond and react in a fashion similar to nitrogen, which is located just above it in the fifth column. Indeed, phosphines - phosphorus analogs of amines - are commonly used in the organic laboratory.
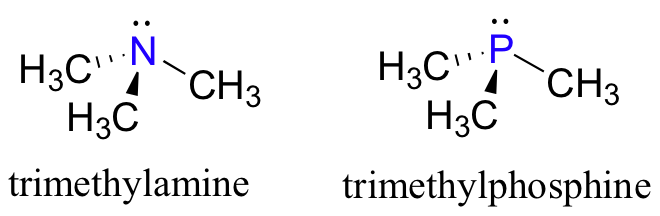
Just like in amines, the central phosphorus atom in a phosphine is sp3 hybridized, with a lone pair of electrons occupying one of the four sp3 hybrid orbitals. In the case of phosphines, however, the sp3 orbitals are hybrids of a single 3s orbital and three 3p orbitals, rather than 2s and 2p orbitals as in amines.

In biological molecules, the most important form of phosphorus is not phosphine, but the phosphoryl group. The four oxygen substituents in phosphoryl groups are arranged about the central phosphorus atom with tetrahedral geometry, however there are a total of five bonds to phosphorus - four s igmabonds and one delocalized pi bond. The reason that phosphorus can break the 'octet rule' is that it is on the third row of the periodic table, and thus has d orbitals available for bonding. In the hybrid orbital picture for phosphate ion (PO4 3-), a single 3s and three 3p orbitals combine to form four sp3 hybrid orbitals with tetrahedral geometry. Four of the five valance electrons on phosphorus occupy sp3 orbitals, and the fifth occupies an unhybridized d orbital.
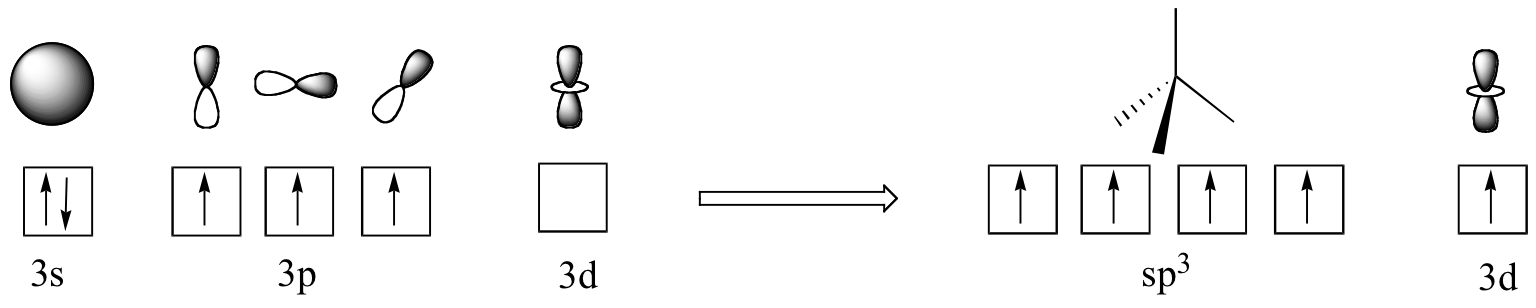
The phosphorus is thus able to form five bonding interactions, rather than three as is phosphine. The four sp3 orbitals on phosphorus are each able to overlap with an sp2 orbital on an oxygen atom (forming a tetrahedral framework), while a delocalized fifth bond (a pi bond) is formed by side-by-side overlap of the d orbital on phosphorus with 2p orbitals on the oxygens.
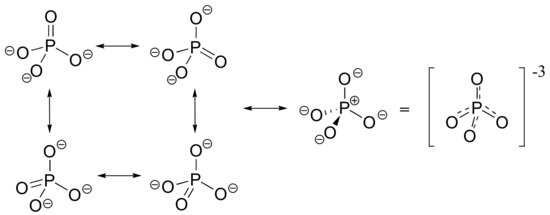
The -3 charge on a fully deprotonated phosphate ion is spread evenly over the four oxygens, and each phosphorus-oxygen bond can be considered have some double bond character.
In phosphate esters, diesters, and anhydrides the pi bonding is delocalized primarily over the non-bridging bonds, while the bridging bonds have mainly single-bond character. In a phosphate diester, for example, the two non-bridging oxygens share a -1 charge, as illustrated by the two major resonance contributors. In the resonance contributors in which the bridging oxygens are shown as double bonds, there is an additional separation of charge - thus these contributors are minor and make a relatively unimportant contribution to the overall bonding picture.
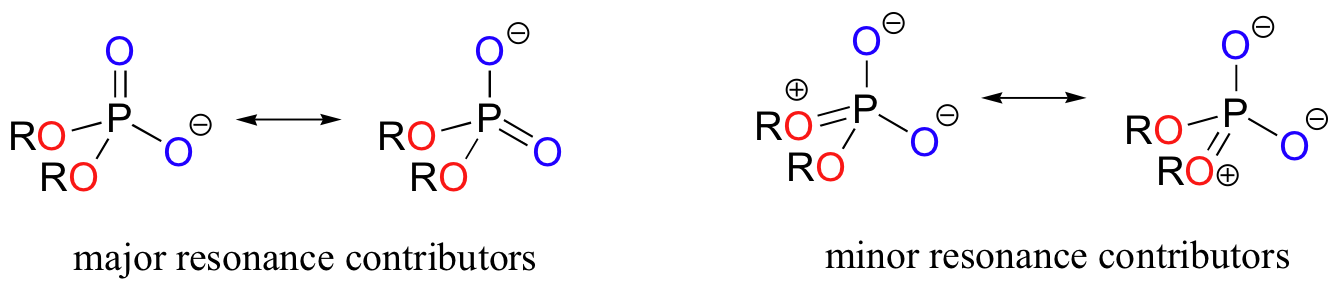

Exercise 10.1: Draw all of the resonance structures showing the delocalization of charge on a (fully deprotonated) organic monophosphate. If a 'bond order' of 1.0 is a single bond, and a bond order of 2.0 is a double bond, what is the approximate bond order of bridging and non-bridging P-O bonds?
Throughout this chapter, phosphoryl groups are usually drawn without attempting to show tetrahedral geometry, and π bonds and negative charges will often be shown as localized to one position. This is done for the sake of simplification - however it is important always to remember that the phosphoryl group is really tetrahedral, and that the pi electrons are delocalized over the non-bridging bonds.
10.1D: Phosphoryl transfer reactions - the general picture
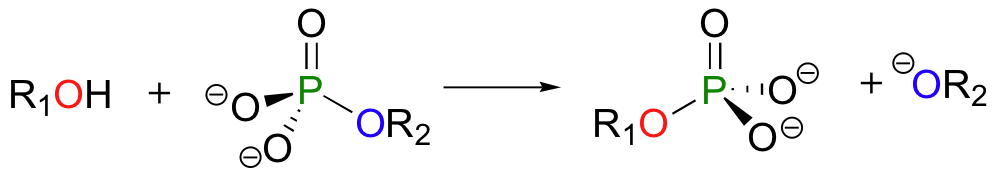
In a phosphoryl transfer reaction, a phosphoryl group is transferred from a phosphoryl group donor molecule (designated 'R2' in the figure below) to a phosphoryl group acceptor (designated 'R1'). The figure below illustrates the transfer of a single phosphate group from R2 to R1:
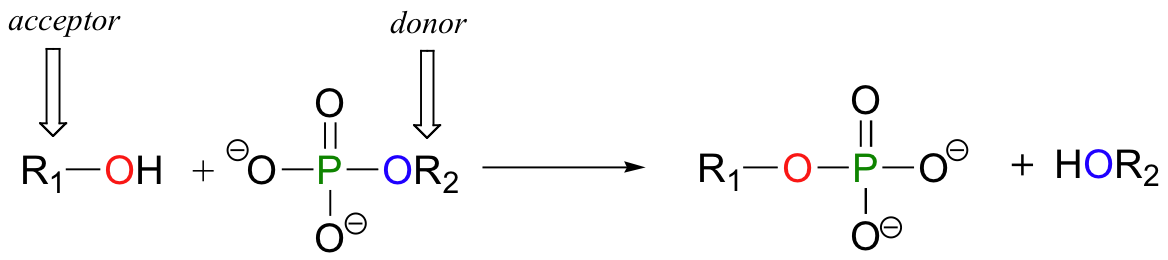
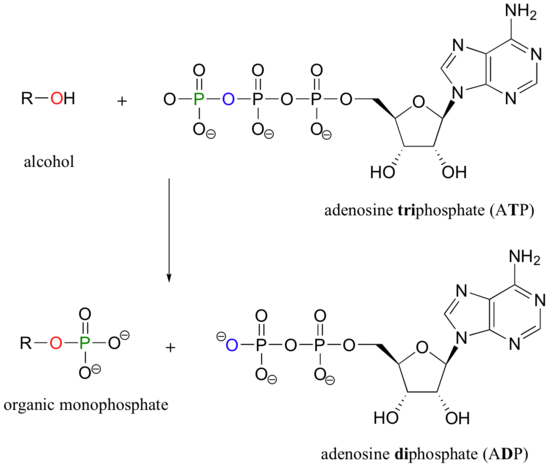
An alcohol, for example, can be transformed into an organic monophosphate by accepting one of the phosphate groups of ATP (we'll learn more about ATP soon).
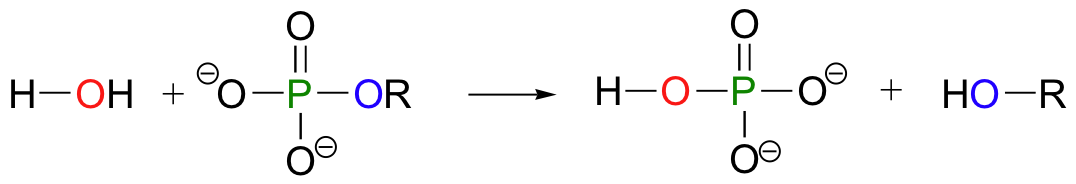
An organic monophosphate is converted back to an alcohol when it transfers its phosphate group to a water molecule:
In the course of this chapter, we will see many other types of phosphoryl transfer reactions, but they all can be described by essentially the same mechanism.
Let's look more closely at the phosphoryl transfer reaction mechanism, using as an example the transfer of a phosphate group from adenosine triphosphate (ATP) to the C6 hydroxyl group of glucose, a reaction catalyzed by glucose kinase.
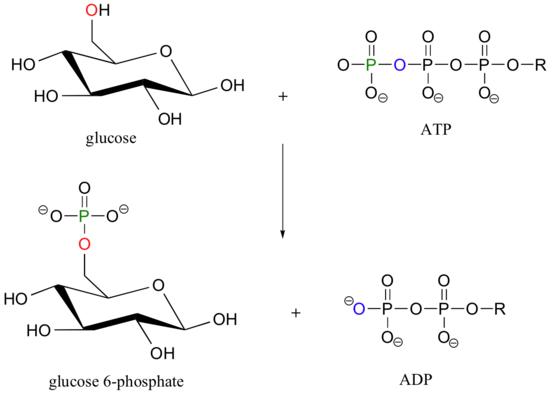
To simplify things in the mechanistic discussion to come, we'll abbreviate this reaction as:
One very important aspect of biological phosphoryl transfer reactions is that the electrophilicity of the phosphorus atom is enhanced by the Lewis acid (electron-accepting) effect of one or more magnesium ions. Phosphoryl transfer-catalyzing enzymes bind Mg2+ ions in such a way that they can interact with non-bridging phosphoryl oxygens on the substrate.
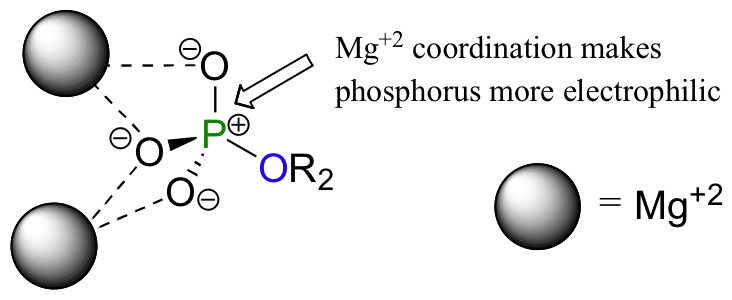
The positively charged metal ions stabilize negative charge on the oxygen atoms, which has the effect of increasing the dipole moment of the phosphorus-oxygen bond. The phosphorus therefore has a larger partial positive charge, which makes it a better electrophile.
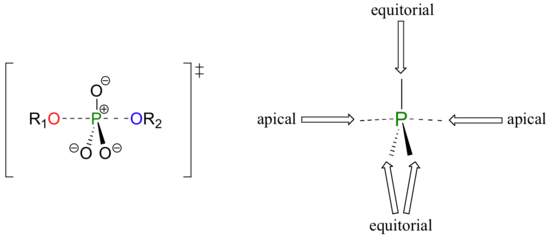
A phosphoryl transfer reaction is very much like a SN2 reaction at a carbon. Just like in an SN2 reaction, the nucleophile in a phosphoryl transfer approaches the electrophilic center from the backside, opposite the leaving group. As the nucleophile gets closer and the leaving group begins its departure, the bonding geometry at the phosphorus atom changes from tetrahedral to trigonal bipyramidal at the transition state. As the phosphorus-nucleophile bond gets shorter and the phosphorus-leaving group bond grows longer, the bonding picture around the phosphorus atom returns to its original tetrahedral state, but the stereochemical configuration has been 'flipped', or inverted.
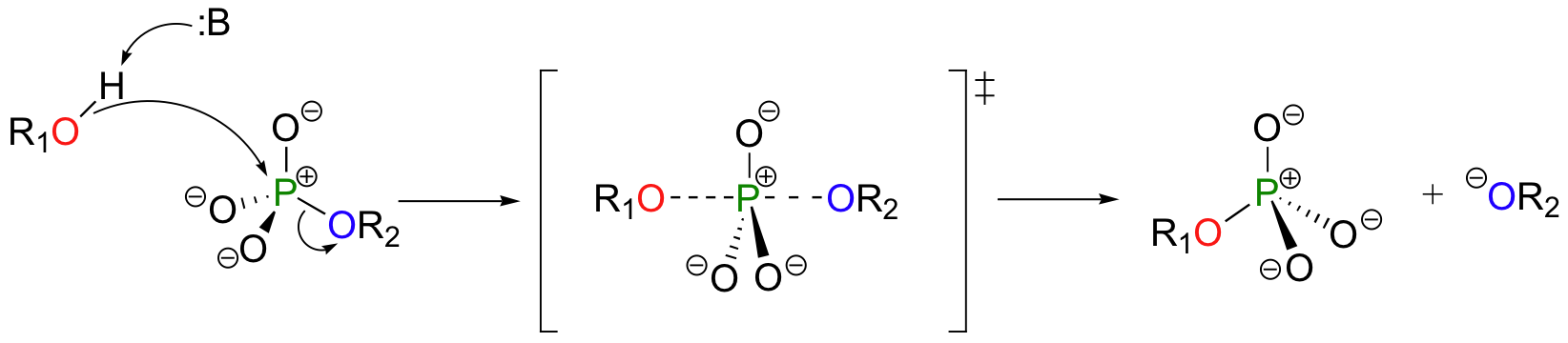
In the trigonal bipyramidal transition state, the five substituents are not equivalent: the three non-bridging oxygens are said to be equatorial (forming the base of a trigonal bipyramid), while the nucleophile and the leaving group are said to be apical (occupying the tips of the two pyramids).
Although stereochemical inversion in phosphoryl transfer reactions is predicted by theory, the fact that phosphoryl groups are achiral made it impossible to observe the phenomenon directly until 1978, when a group of researchers was able to synthesize organic phosphate esters in which stable oxygen isotopes 17O and 18O were specifically incorporated. This created a chiral phosphate center.

Subsequent experiments with phosphoryl transfer-catalyzing enzymes confirmed that these reactions proceed with stereochemical inversion. (Nature 1978 275, 564; Ann Rev Biochem 1980 49, 877).
10.1E: Phosphoryl transfer reactions - concerted, addition-elimination, or dissociative?
In the above discussion, the phosphoryl transfer reaction mechanism was depicted as passing through a concerted SN2-like transition state, with both apical bonds in some stage of breaking or forming at the top of the 'energy hill':
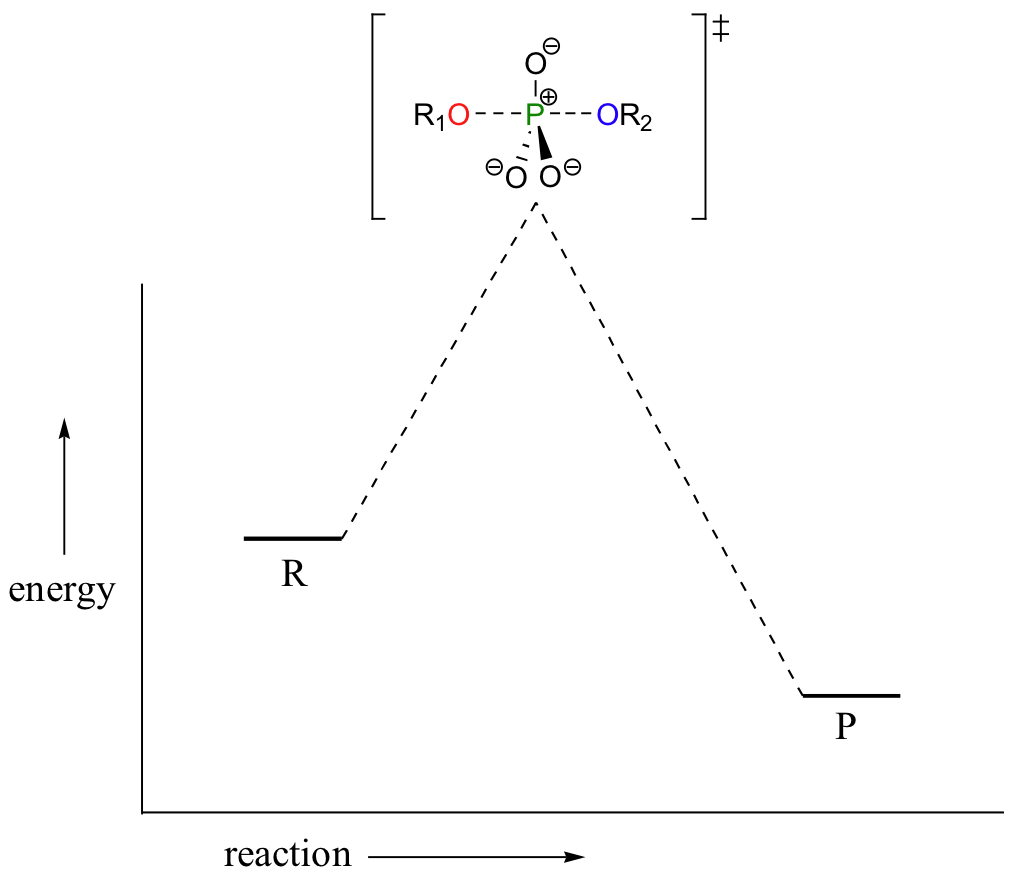
This is not the only mechanism that has been proposed for these reactions - in fact, two other possible mechanisms have been suggested.
In an alternative two-step mechanistic model, the nucleophile could attack first, forming a pentavalent, trigonal bipyramidal intermediate, (as apposed to a pentavalent transition state). The reaction is completed when the leaving group is expelled. The intermediate species would occupy an energy valley between the two transition states.
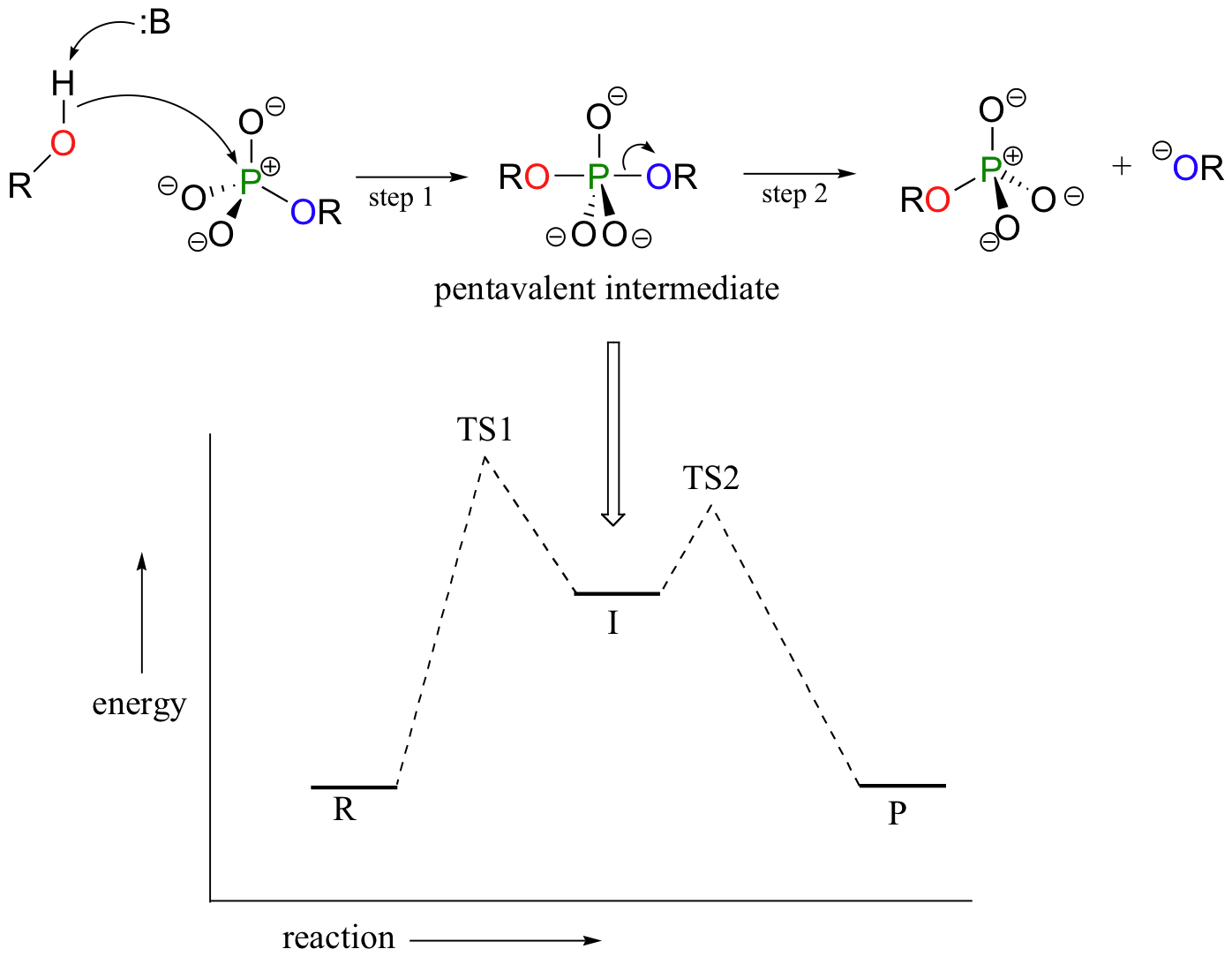
This is often referred to as an 'addition-elimination' mechanism - the nucleophile adds to the phosphate first, forming a pentavalent intermediate, and then the leaving group is eliminated.
A pentavalent intermediate is not possible for an SN2 reaction at a carbon center, because carbon, as a second-row element, does not have any d orbitals and cannot form five bonds. Phosphorus, on the other hand, is a third-row element and is quite capable of forming more than four bonds. Phosphorus pentachloride, after all, is a stable compound that has five bonds to chlorine arranged in trigonal bipyramidal geometry around the central phosphorus.
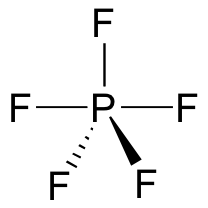
The phosphorus atom in PCl5 (and in the hypothetical pentavalent intermediate pictured above) is considered to be sp3d hybridized:

There is a third possibility: the reaction could proceed in a dissociative, SN1-like manner. In this model, the phosphorus-leaving group bond breaks first, resulting in a 'metaphosphate' intermediate. This intermediate, which corresponds to the carbocation intermediate in an SN1 reaction, is then attacked by the nucleophile to form the reaction product.

So what is the actual mechanism for a phosphoryl transfer reaction - concerted, addition-elimination, or dissociative? Chemists love to investigate and argue about questions like this! Just like with the SN1/SN2 argument discussed in the previous chapter, it really boils down to one question. Which happens first, bond-forming or bond-breaking - or do these two events occur at the same time? From the evidence accumulated to date, it appears that enzymatic phosphoryl transfer reactions may occur by all three mechanisms - and often somewhere in between - depending on the nature of the nucleophile, the electrophile, and the leaving group, as well as on the active-site architecture of the enzyme catalyzing the reaction. Although it is thought that many phosphoryl transfer reactions, both enzymatic and non-enzymatic, proceed with some degree of dissociative (SN1-like) character, there is not yet a clear understanding of exactly what happens between starting compound and product. Considering the importance of phosphoryl transfer reactions in metabolic pathways, this area is clearly a very promising one for further investigation. (FASEB J. 1995 9, 1585; Trends Biochem Sci. 2004 29, 495).
For the sake of simplicity and clarity, phosphoryl transfers in this text will generally be depicted as concerted, SN2-like reactions, in two dimensions, with a localized double bond. but you should be always keep in mind the existence of a pentavalent, trigonal bipyramidal transition state/intermediate. Also, be aware that in other books and articles these reactions may be drawn somewhat differently.