
13.3: Aldol reactions
- Page ID
- 979

We come now to one of the most important mechanism types in metabolism: the carbon-carbon bond-forming 'aldol' condensation reaction.
13.3A: The general mechanism for an aldol reaction
Consider the potential pathways available to a reactive enolate intermediate once the alpha-proton has been abstracted. The oxygen, which bears most of the negative charge, could act as a base, and the result would be an enol.

Alternatively, the enolate carbon, which bears a degree of negative charge, could act as a base, which is simply the reverse of the initial deprotonation step that formed the enolate in the first place.

In both of these cases, the electron-poor species attacked by the enolate is an acidic proton. What if the electron-poor species - the electrophile - is not a proton but a carbonyl carbon? In other words, what if the enolate acts not as a base but rather as a nucleophile in a carbonyl addition (chapter 11) reaction? One possibility is that the enolate oxygen could be the nucleophile in a hemiketal-forming reaction:
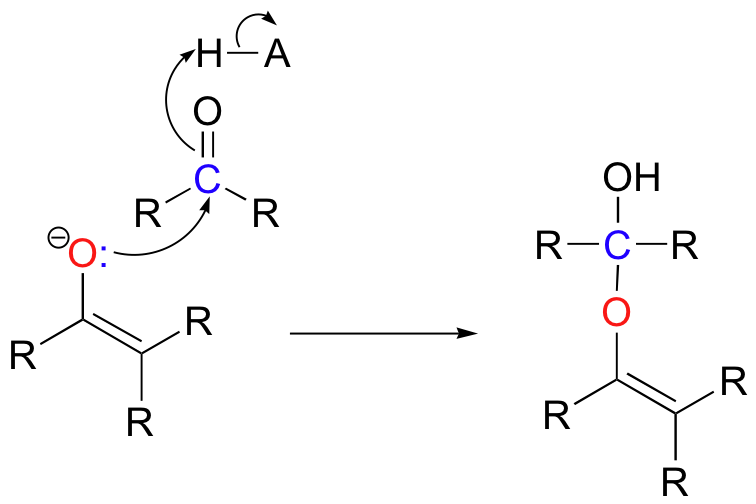
Although you can find examples of this type of reaction in biochemistry, it is far more common for the enolate carbon to be the nucleophile, attacking an electrophilic carbonyl to form a new carbon-carbon bond.
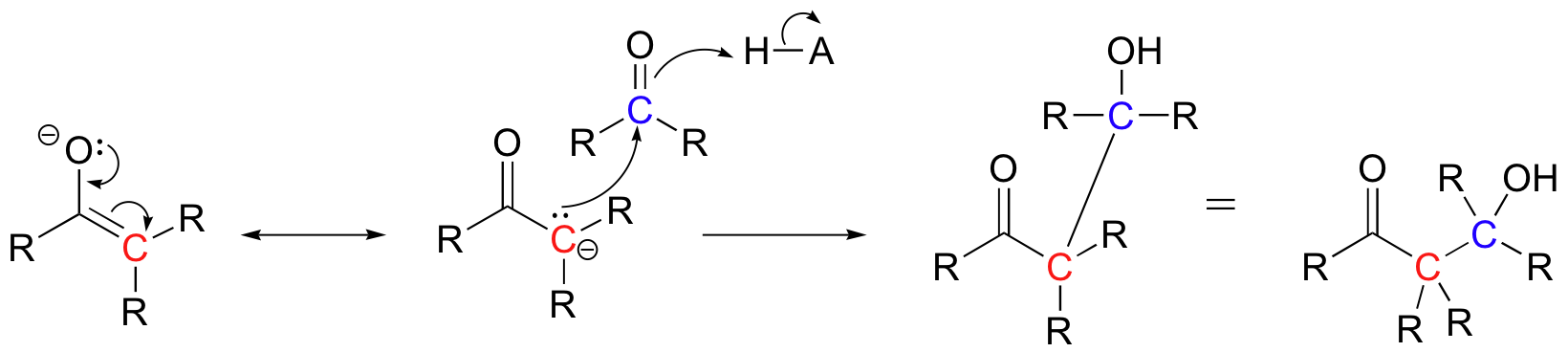
Historically, the first examples of this mechanism type to be studied involved the non-enzymatic reaction of an aldehyde with itself (a so-called 'self-condensation' reaction, where 'condensation' means the formation of one larger molecule from two smaller ones).
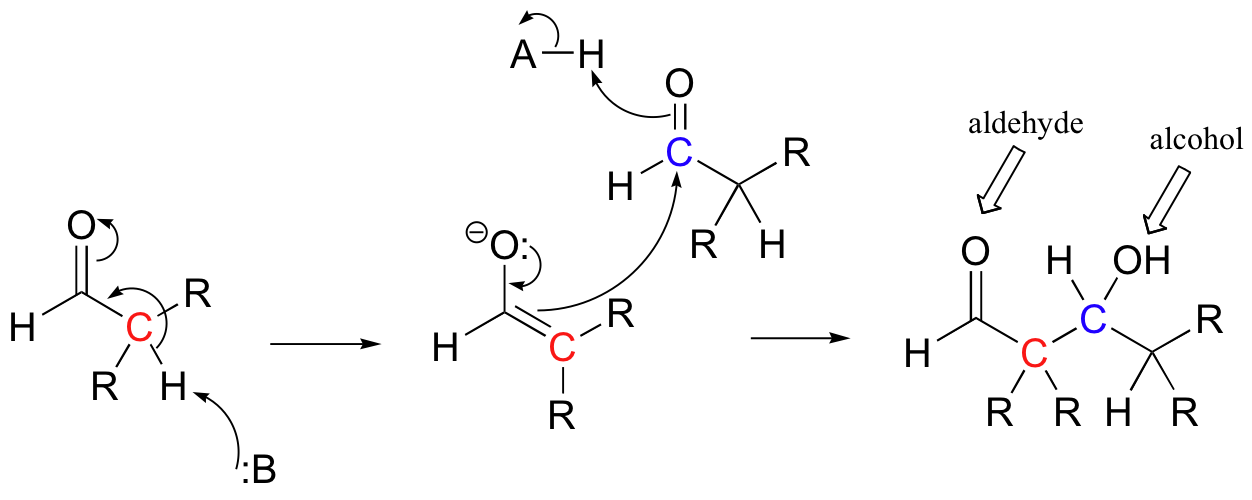
Because the resulting product contained both an aldehyde and an alcohol functional group, the reaction was termed an 'aldol condensation', a name that has become standard for reactions of this type, whether or not an aldehyde is involved.
The enzymes that catalyze aldol reactions are called, not surprisingly, 'aldolases'.
13.3B: Typical aldolase reactions - three variations on a theme
The first step in an aldolase reaction is the deprotonation of an alpha-carbon to generate a nucleophilic carbanion. Nature has evolved several distinct strategies to stabilize the intermediate that results. Some aldolases use a metal ion to stabilize the negative charge on an enolate intermediate, while others catalyze reactions that proceed through neutral Schiff base or enol intermediates.
Let's examine first a reaction catalyzed by a so-called 'Class II' aldolase, in which a metal cation - generally Zn2+ - bound in the active site serves to stabilize the negative charge on an enolate intermediate. Fructose 1,6-bisphosphate aldolase is an enzyme that participates in both the glycolytic (sugar burning) and gluconeogenesis (sugar building) biochemical pathways. For now, we will concentrate on its role in the gluconeogenesis pathway, but we will see it again later in its glycolytic role. The reaction catalyzed by fructose 1,6-bisphosphate aldolase is a condensation between two 3-carbon sugars, glyceraldehyde-3-phosphate (GAP) and dihydroxyacetone phosphate (DHAP), forming a six-carbon product (which leads, after three more enzymatic steps, to glucose).
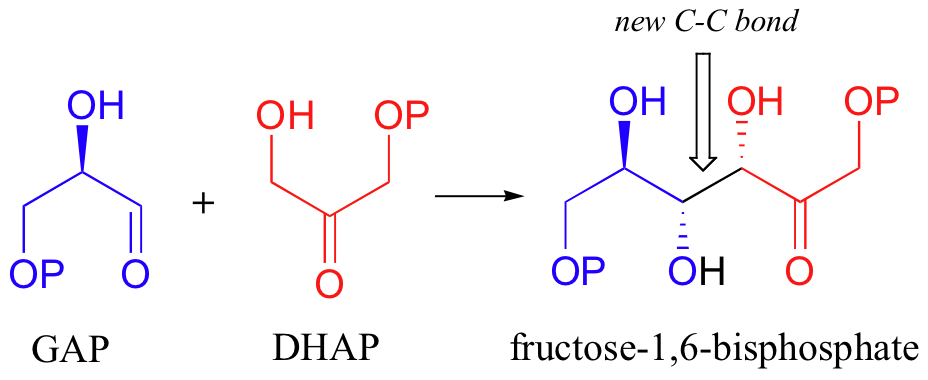
In the first step of the condensation, an alpha-carbon on DHAP is deprotonated, leading to an enolate intermediate. The strategy used to stabilize this key intermediate is to coordinate the negatively-charged enolate oxygen to an enzyme-bound zinc cation.
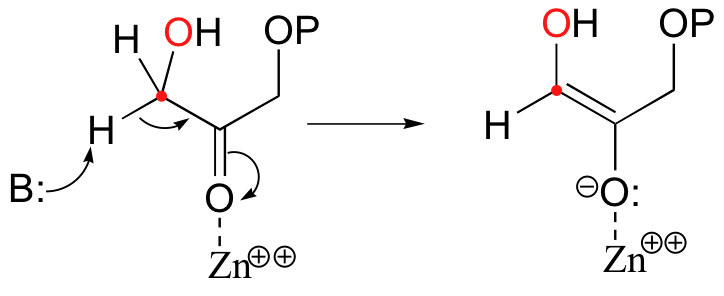
Next, the deprotonated a-carbon attacks the carbonyl carbon of GAP in a nucleophilic addition reaction, and protonation of the resulting alcohol leads directly to the fructose 1,6-bisphosphate product.
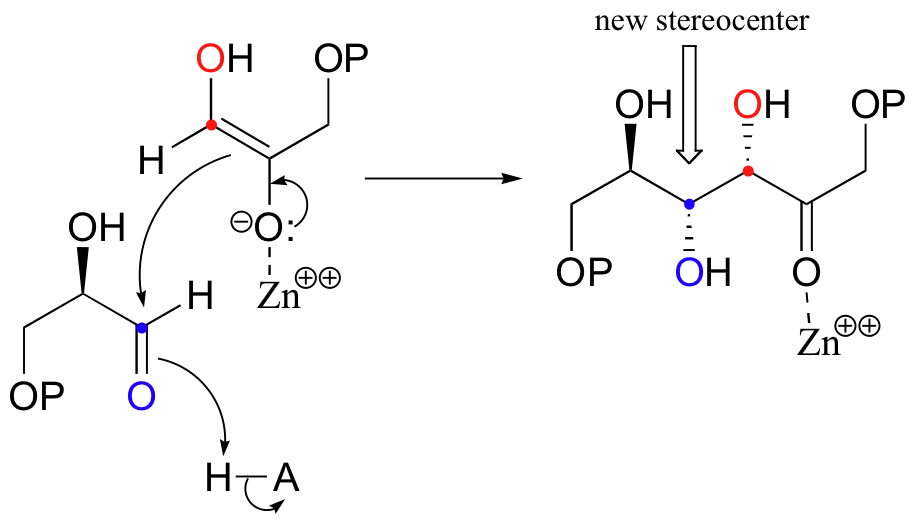
As with many other nucleophilic carbonyl addition reactions, a new stereocenter is created in this reaction, as a planar, achiral carbonyl group is converted to a tetrahedral, chiral alcohol. The enzyme-catalyzed reaction, not surprisingly, is completely stereospecific: the DHAP substrate is positioned in the active site so as to attack the re (front)face of the GAP carbonyl group, leading to the R configuration at the new stereocenter.
Interestingly, it appears that in bacteria, the fructose bisphosphate aldolase enzyme evolved separately from the corresponding enzyme in plants and animals. In plants and animals, the same aldol condensation reaction is carried out by a significantly different mechanism, in which the key intermediate is not a zinc-stabilized enolate but an enamine. The nucleophilic substrate (DHAP) is first linked to the enzyme through the formation of an imine (also known as a Schiff base, section 11.6) with a lysine residue in the active site.
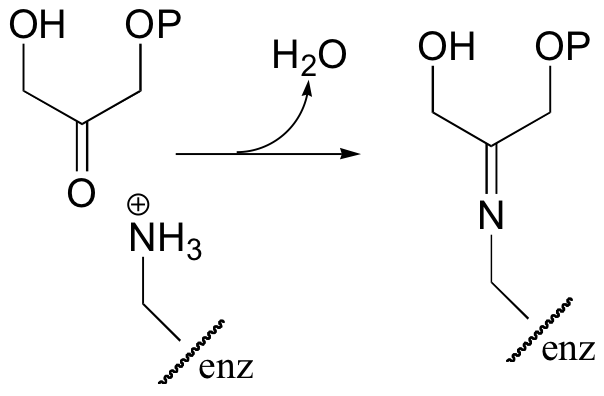
The alpha-proton is then abstracted by an active site base to form an enamine.
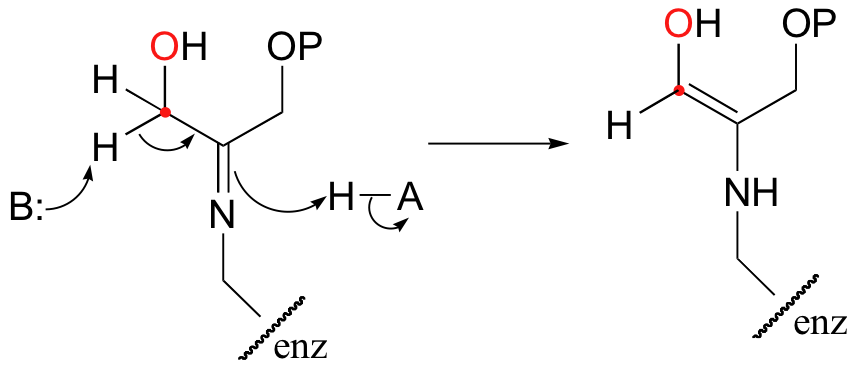
In the next step, the alpha-carbon attacks the carbonyl carbon of GAP, and the new carbon-carbon bond is formed. In order to release the product from the enzyme active site and free the enzyme to catalyze another reaction, the imine is hydrolyzed back to a ketone group.

There are many more examples of 'Class I' aldolase reactions in which the key intermediate is a lysine-linked imine. Many bacteria are able to incorporate formaldehyde, a toxic compound, into carbohydrate metabolism by condensing it with ribulose monophosphate. The reaction proceeds through imine and enamine intermediates.

a) Propose a complete mechanism for the condensation reaction shown above.
b) Propose a complete mechanism for the conversion of hexulose-6-phosphate (formed from the condensation of ribulose-5-phosphate and formaldehyde) into fructose-6-phosphate.
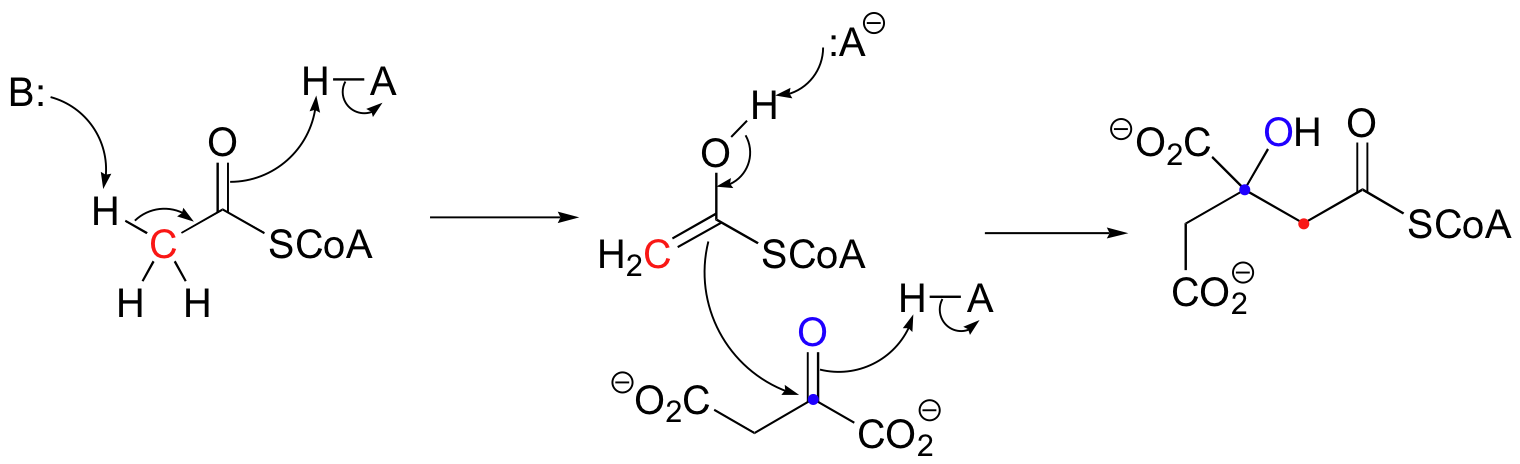
Notice that in this aldol reaction, the nucleophilic intermediate is stabilized by protonation, rather than by formation of an imine (as in the Class I aldolases) or by a metal ion (as in the Class II aldolases).
13.3C: Going backwards: the retroaldol reaction
Although aldol reactions play a very important role in the formation of new carbon-carbon bonds in metabolic pathways, it is important to emphasize that they are also highly reversible: in most cases, the energy level of starting compounds and products are very close. This means that, depending on metabolic conditions, aldolases can also catalyze retro-aldol reactions (the reverse of aldol condensations, in which carbon-carbon bonds are broken). Recall that fructose 1,6-bisphosphate aldolase (section 13.3B) is active in the direction of sugar breakdown (glycolysis) as well as sugar synthesis (gluconeogenesis). In the glycolytic direction, the enzyme catalyzes - either by zinc cation or by imine/enamine mechanisms, depending on the organism - the retro-aldol cleavage of fructose bisphosphate into DHAP and GAP.

The mechanism is the exact reverse of the condensation reaction. Shown below is the mechanism for a Zn2+ - dependent (Type II) retroaldol cleavage. Notice that in the retroaldol reaction, the enolate intermediate is the leaving group, rather than the nucleophile.
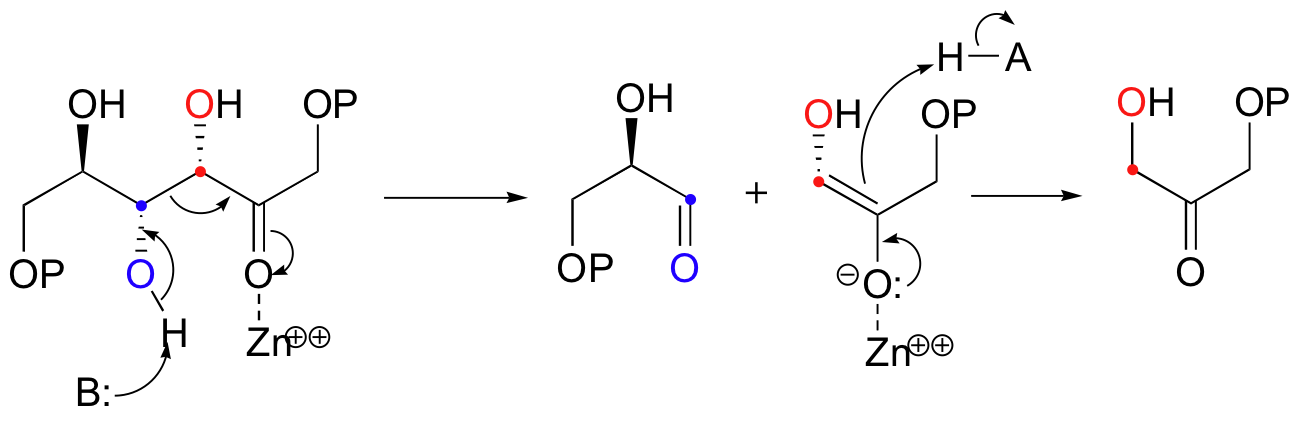
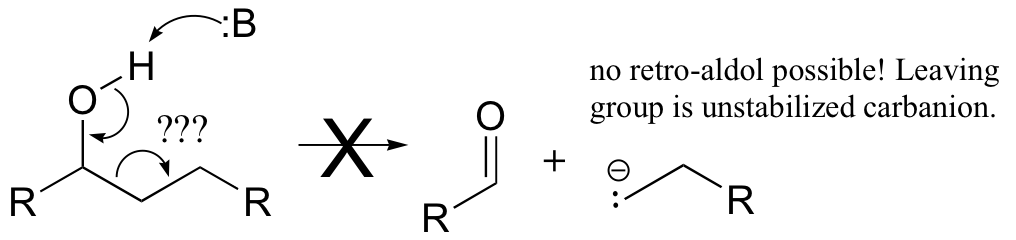
The key thing to keep in mind when looking for a possible retro-aldol mechanism is that, when the carbon-carbon bond breaks, the electrons must have some place to go, where they will be stabilized by resonance. Generally, this means that there must be a carbonyl or imine group on the next carbon. If there is no adjacent carbonyl or imine group, the carbon-carbon bond is not free to break.
Here are two more examples of retro-aldol reactions. Bacterial carbohydrate metabolism involves this reversible, class I retro-aldol cleavage: (Proc. Natl. Acad. Sci 2001, 98, 3679).
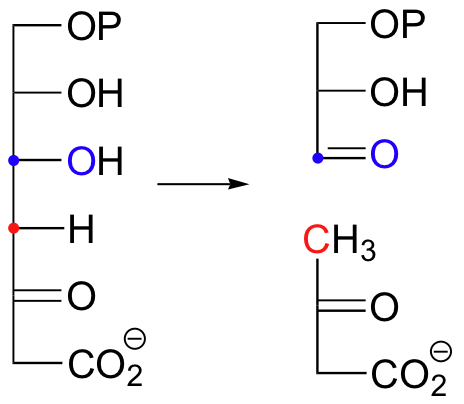
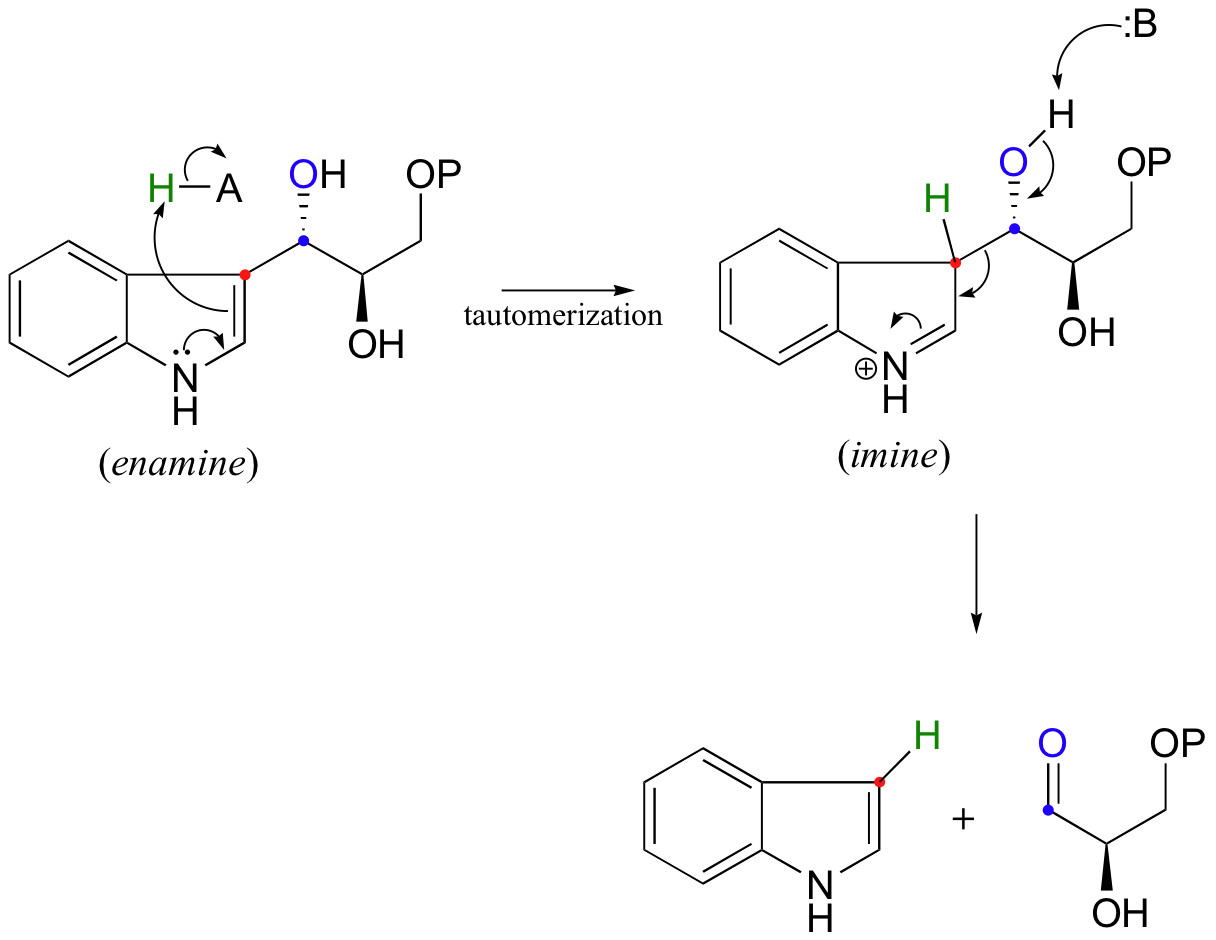
Look carefully at this reaction - how is the leaving group stabilized? There is an imine group involved, but no participation by an enzymatic lysine. The imine is 'built into' the starting compound, available from the initial tautomerization of the cyclic enamine group in indole-3-glycerol phosphate.
13.3D: Going both ways: transaldolase
An enzyme called transaldolase, which is part of the 'pentose phosphate pathway' of carbohydrate metabolism, catalyzes an interesting combination of class I aldol and retro-aldol reactions. The overall reaction, which can proceed in either direction depending on metabolic requirements, converts 3- and 7-carbon sugars into 6- and 4-carbon sugars. Essentially, a 3-carbon unit breaks off from a ketone sugar (ketose) and then is condensed directly with an aldehyde sugar (aldose).

Let's follow the progress of the reaction in the left-to-right direction as depicted above. Because this is a class I aldolase, the first step is the formation of an imine linkage between the ketone carbon of fructose-6-phosphate (F6P) and a lysine group from the enzyme. The enzyme-substrate adduct then undergoes a retro-aldol step to free glyceralde-3-phosphate (GAP), which leaves the active site.

The second substrate, erythrose 4-phosphate (E4P), enters the active site, and an aldol condensation occurs between E4P and the 3-carbon fragment remaining from the cleavage of fructose-6-phosphate.

The final step is hydrolysis of the imine and subsequent dissociation of sedoheptulose 7-phosphate from the active site.