
13.5: Carboxylation and decarboxylation reactions
- Page ID
- 981

13.5A: The metabolic context of carboxylation and decarboxylation
Some of the most important carbon-carbon bond-forming and bond-breaking processes in biological chemistry involve the gain or loss, by an organic molecule, of a single carbon in the form of CO2. You undoubtedly have seen this chemical equation before in an introductory biology or chemistry class:
6CO2 + 6H2O + energy → C6H12O6 + 6O2
This of course represents the photosynthetic process, by which plants (and some bacteria) harness energy from sunlight to build glucose from individual carbon dioxide molecules. The key chemical step in which carbon dioxide is 'fixed' (in other words, condensed with an existing organic molecule) is called a carboxylation reaction. It is catalyzed by the enzyme ribulose 1,5-bisphosphate carboxylase, commonly known as Rubisco, in the 'Calvin cycle' of carbon fixation.
The reverse chemical equation is also probably familiar to you:
C6H12O6 + 6O2 → 6CO2 + 6H2O + energy
This equation expresses what happens in respiration: the oxidative breakdown of glucose to form carbon dioxide, water, and energy. In the course of this transformation, each of the carbon atoms of glucose is eventually converted to individual CO2 molecules. The actual chemical step by which a carbon atom, in the form of carbon dioxide, breaks off from a larger organic molecule is called a decarboxylation reaction. The key decarboxylation steps in the conversion of glucose to carbon dioxide occur in the citric acid (Krebs) cycle and the pentose phosphate pathway.
Let's now look at the organic mechanisms of some carboxylation and decarboxylation reactions.
13.5B: The carboxylation mechanism of Rubisco, the carbon fixing enzyme
Carboxylation reactions are essentially just aldol condensations, except that the carbonyl electrophile is CO2 rather than a ketone or aldehyde. The mechanism for Rubisco, the key carbon-fixing enzyme in plants and photosynthetic bacteria (and the most abundant enzyme on earth!), is shown below. Magnesium ion plays a key charge-stabilizing role throughout the reaction. Step 1, not surprisingly, is deprotonation of an alpha-carbon to form an enolate.

Step 2 is simply an intramolecular proton transfer, which has the effect of creating a different enolate intermediate and making C2 into the nucleophile for an aldol-like attack on CO2 (step 3). Carbon dioxide has now been 'fixed' into organic form - it has become a carboxylate group on a six-carbon sugar derivative.
To follow the Rubisco mechanism through to its endpoint:

Step 4 is a retro-Claisen mechanism, with a water nucleophile and enolate leaving group. After protonation of this enolate, we are left with two molecules of 3-phosphoglycerate, which are incorporated into the 'gluconeogenesis' pathway of glucose synthesis.
| Example 13.8 |
|---|
| Propose a complete mechanism for the retro-Claisen reaction in the figure above. |
13.5C: Decarboxylation
Mechanistically, a decarboxylation has parallels to retro-aldol cleavage reactions:
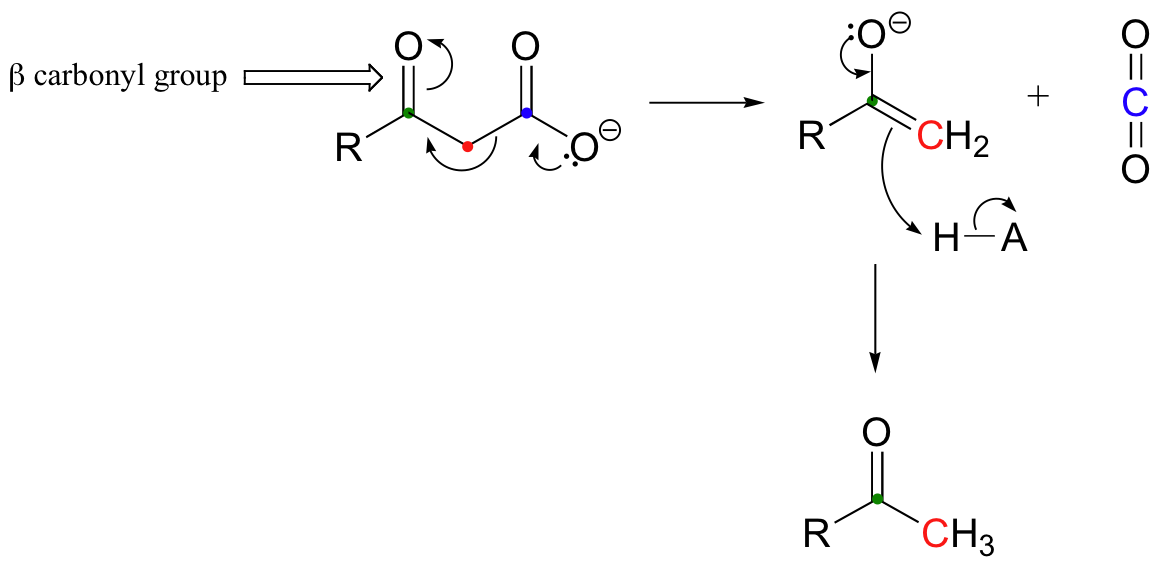
Just as in retro-aldol reactions, the electrons from the broken carbon-carbon bond have to have some place to go - they must, in other words, be stabilized - for the decarboxylation step to take place. Quite often, the electrons are stabilized by the formation of an enolate, as is the case in the general mechanism pictured above. This of course means that a carbonyl group must be positioned beta to (i.e. two carbons down from) the carboxylate carbon. If there is no stable place for the electrons in the carbon-carbon bond to go, then a decarboxylation is very unlikely.
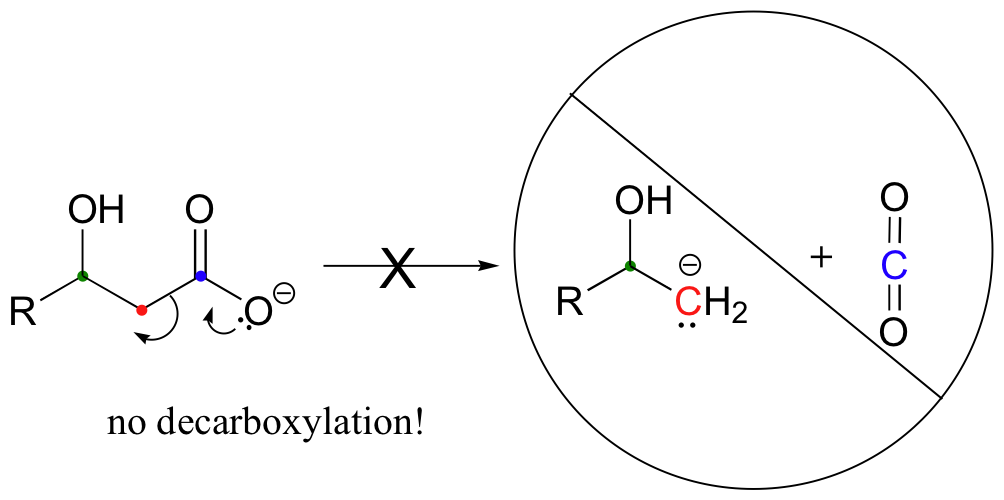
Be especially careful, when drawing decarboxylation mechanisms, to resist the temptation to treat the CO2 molecule as the leaving group:
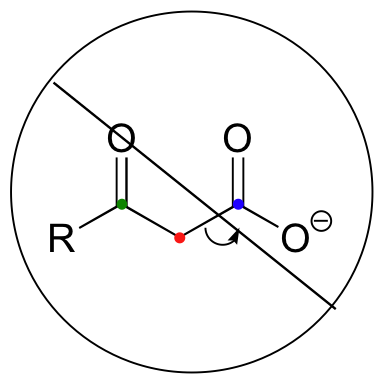
This is not what a decarboxylation looks like! In a decarboxylation step, it is the organic part of the molecule that is, in fact, the leaving group, 'pushed off' by the electrons on the carboxylate.
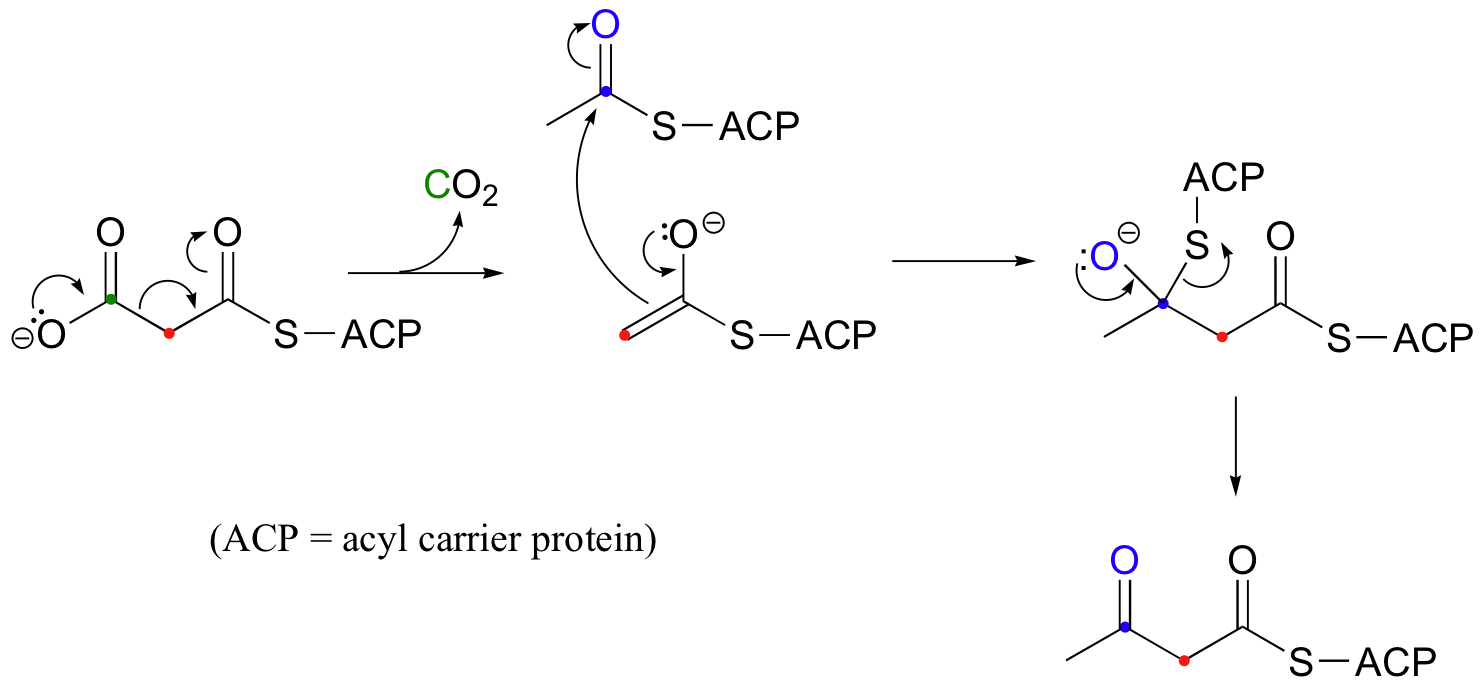
Below are two important key b-carbonyl decarboxylation steps in glucose metabolism, each representing a point at which a carbon derived from glucose is released as CO2.
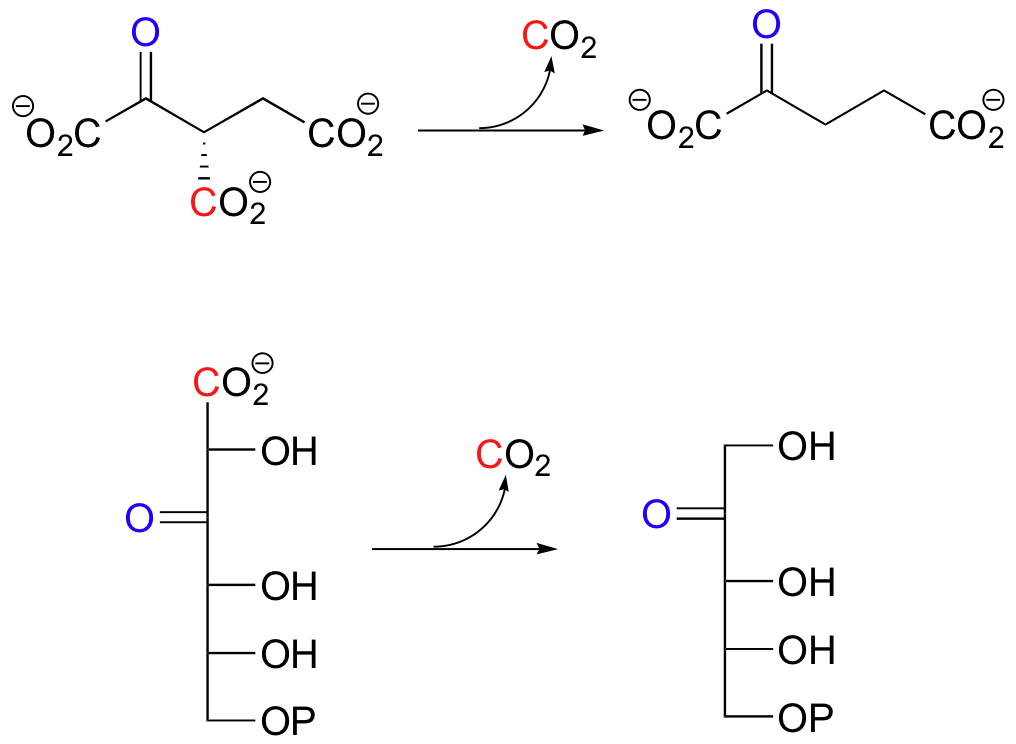
| Example 13.9 |
|---|
| Exercise 13.9: Draw mechanistic arrows showing the first step in each of the reactions shown above. |
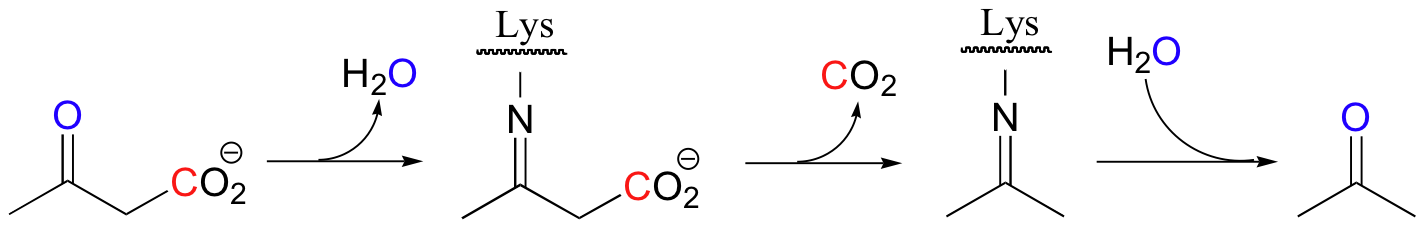
| Example 13.10 |
|---|
| Exercise 13.10: Draw a mechanism for the decarboxylation step in the reaction above. |
An interesting variation on decarboxylation in the synthesis of tyrosine in bacteria is shown below.
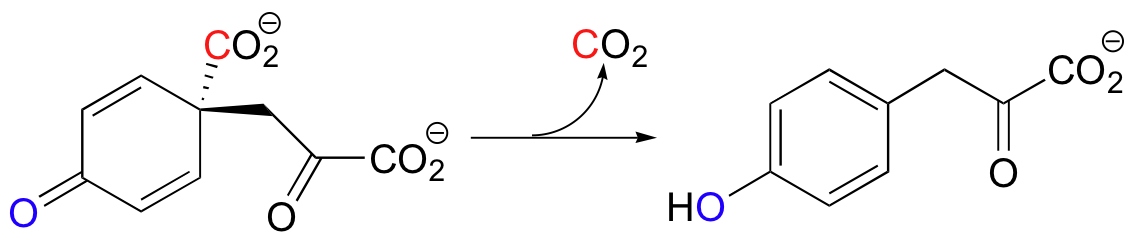
| Example 13.11 |
|---|
| a) Draw a mechanism for the reaction above. b) Draw hypothetical decarboxylation mechanisms showing the formation of alternate products A and B from the same starting compound:
c) How would you describe in words the relationship between the actual product and hypothetical products A and B? Which of the three would you expect to be most thermodynamically stable, and why? |

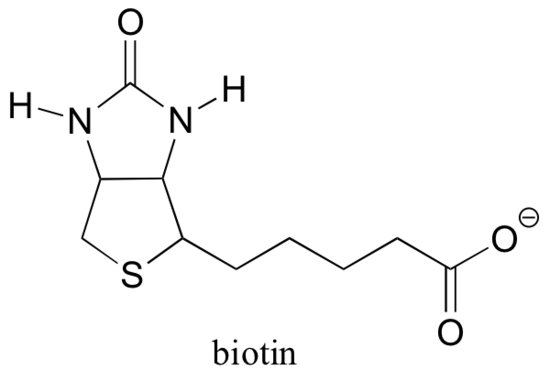
13.5D: Biotin is a CO2-carrying coenzyme
Recall from section 6.5B that many enzymes are dependent upon the assistance of coenzymes, which are small (relative to protein) organic molecules that bind - covalently or non-covalently - in an enzyme's active site and help it to catalyze its reaction. S-adenosylmethionine (SAM, section 9.1A) and ATP (section 10.2) are two examples that we have encountered so far, and we will see several more in the chapters ahead. Although Rubisco (described in part B of this section) is an exception, most enzymes that catalyze carboxylation reactions are dependent upon a coenzyme called biotin, which serves as a temporary carrier of carbon dioxide.
Pyruvate carboxylase, the enzyme catalyzing the first step of the gluconeogensis pathway, is a good example of a biotin-dependent carboxylation reaction. Notice that the CO2 in this reaction is derived from bicarbonate, unlike the Rubisco reaction in which CO2 is 'fixed' directly from the atmosphere.

Biotin is covalently attached to the enzyme through an amide linkage to an active site lysine.
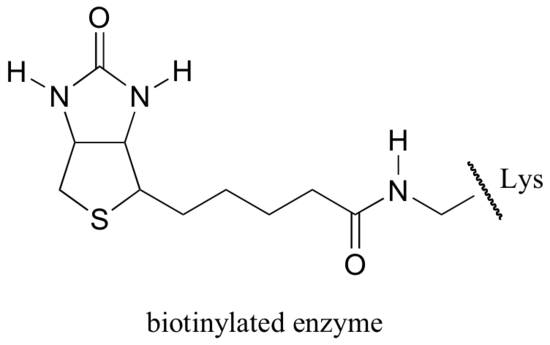
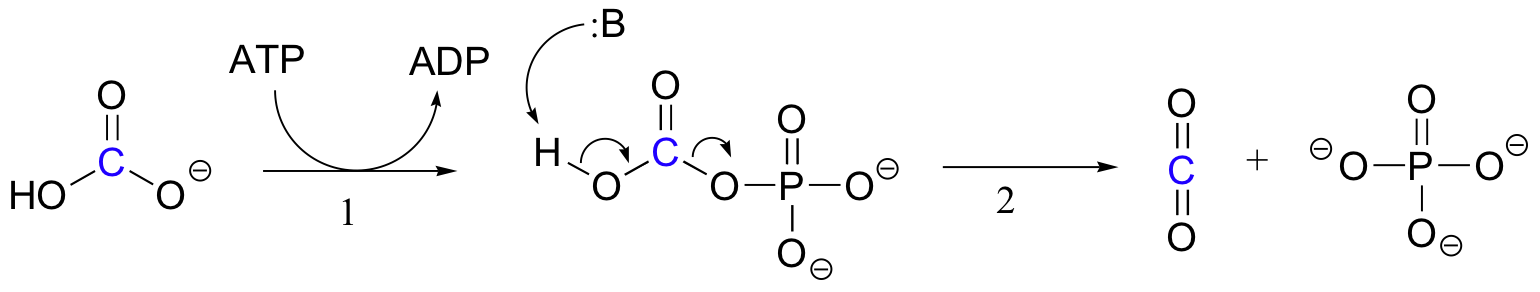
The exact mechanism by which biotin-dependent carboxylation reactions operate is still not completely understood, however the following is a likely picture. First, the bicarbonate ion is phosphorylated by ATP (step 1, see section 10.2), and thus is activated for decarboxylation, which generates free CO2 (step 2).
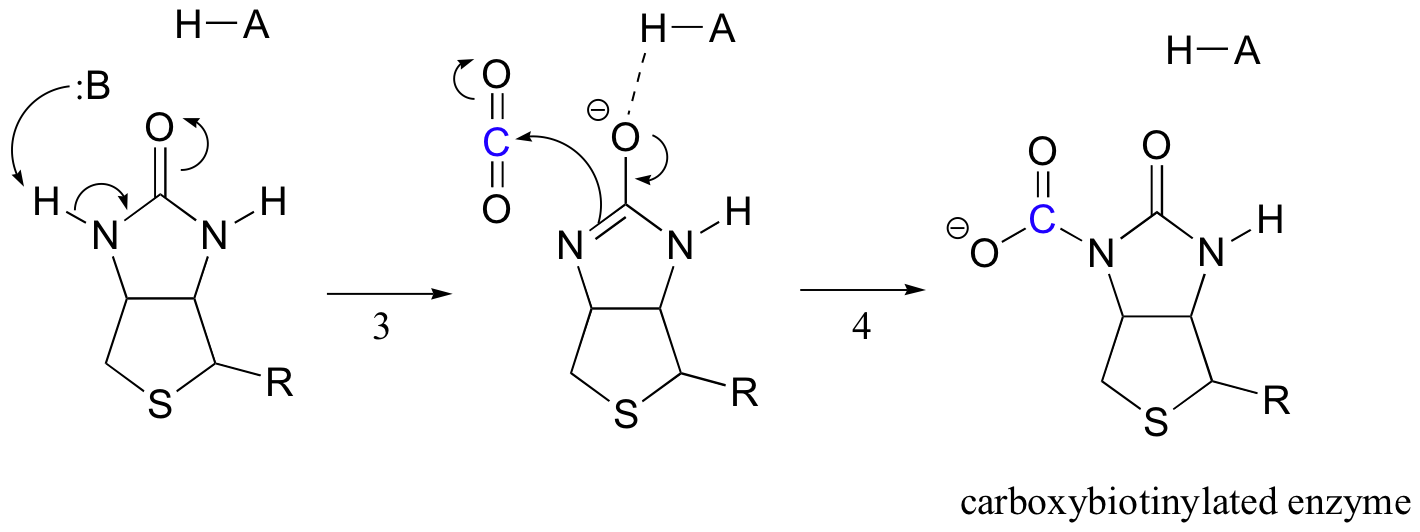
Biotin's job is to hold on to the carbon dioxide molecule until pyruvate comes into the active site. Carboxylation of biotin involves deprotonation of the amide nitrogen to form an enolate-like intermediate (step 3 - amides have a pKa of approximately 17, and this is lowered by the presence of an active site acid near the oxygen). This step is followed by attack of the nucleophilic nitrogen on carbon dioxide to form carboxybiotinylated enzyme (step 4).
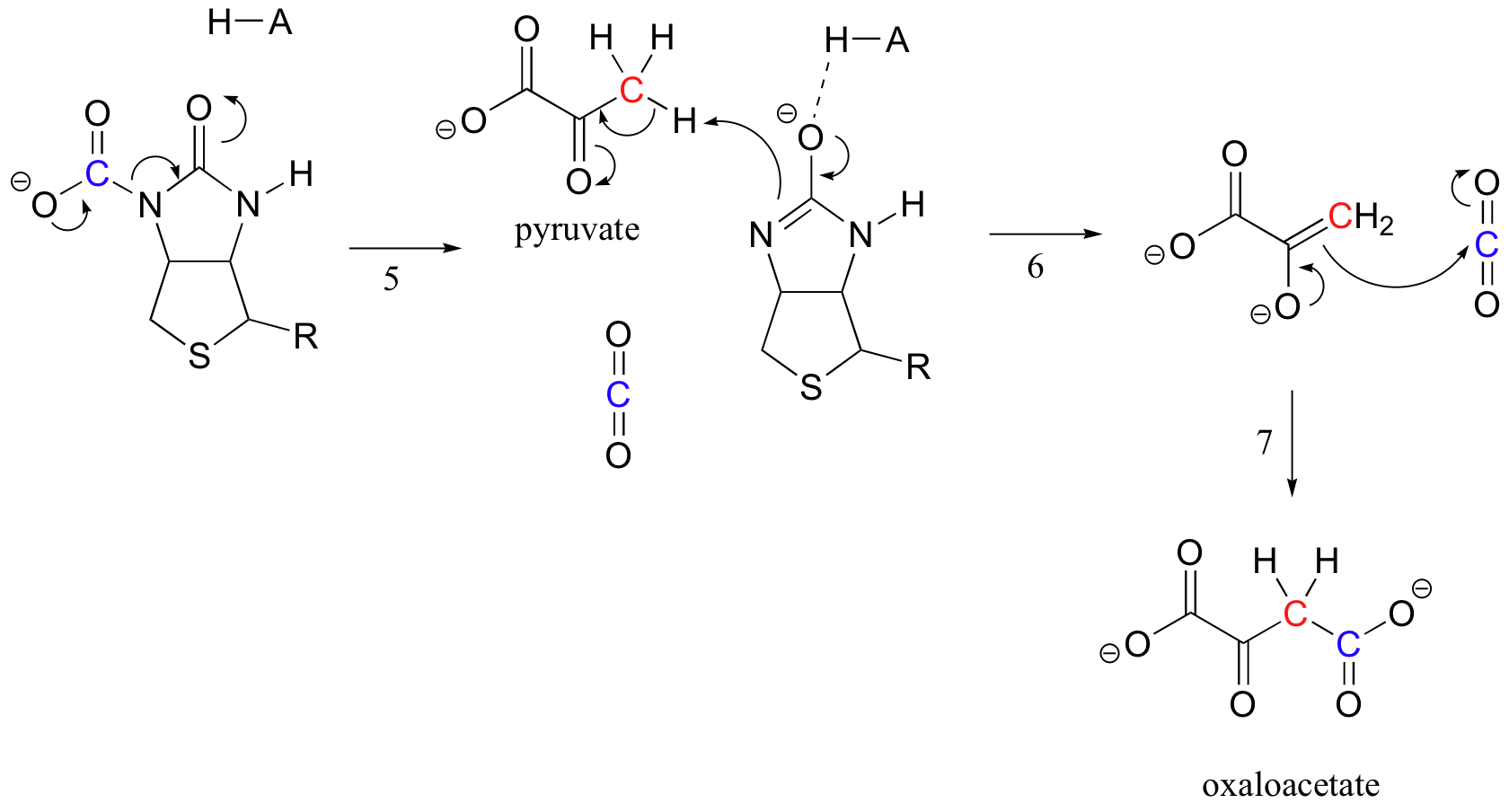
When a pyruvate molecule binds, rearrangement of the active site architecture causes the previous step to go in reverse (step 5), freeing the CO2 and generating a biotin base to deprotonate the alpha-carbon of pyruvate so that it can condense, in an aldol-like fashion, with CO2 to form oxaloacetate (steps 6-7).
If you have studied some biochemistry, you may have heard about biotin in a somewhat different context that what is discussed in this section. A protein called avidin, found in abundance in egg white, binds non-covalently and extremely tightly to biotin (in fact, avidin-biotin is the tightest protein-ligand binding pair known to science). Biochemists often make use of this property by covalently linking a biomolecule of interest to biotin. The 'biotinylated' species can then be easily isolated from a complex mixture by running the mixture through an 'affinity column' that is coated with avidin.