
15.6: Synthetic parallel - electrophilic aromatic substitution in the lab
- Page ID
- 999

15.6A: Friedel-Crafts reactions
Compounds containing aromatic groups are widespread in nature, and for this reason chemists who aim to synthesize naturally occurring compounds in the laboratory often need to introduce substituents to aromatic rings. In the organic synthesis laboratory, electrophilic aromatic substitutions which result in the formation of new carbon-carbon bonds are called ‘Friedel-Crafts’ alkylations and acylations, named for Charles Friedel of France and James Crafts of the United States, who together developed the procedures in 1877. The Friedel-Crafts reactions are an important part of a synthetic chemist's toolbox to this day.
Friedel Crafts reactions, like their biochemical counterparts, require reactive electrophiles with significant carbocation character. One of the most common ways to alkylate an aromatic ring is to use an alkyl chloride electrophile that is activated by the addition of aluminum or iron trichloride. The metal chloride serves as a Lewis acid, accepting electron density from the alkyl chloride. This has the effect of magnifying the carbon-chlorine dipole, making the carbon end more electropositive - and thus more electrophilic – even to the point where the bond breaks and an ion-pair is formed.
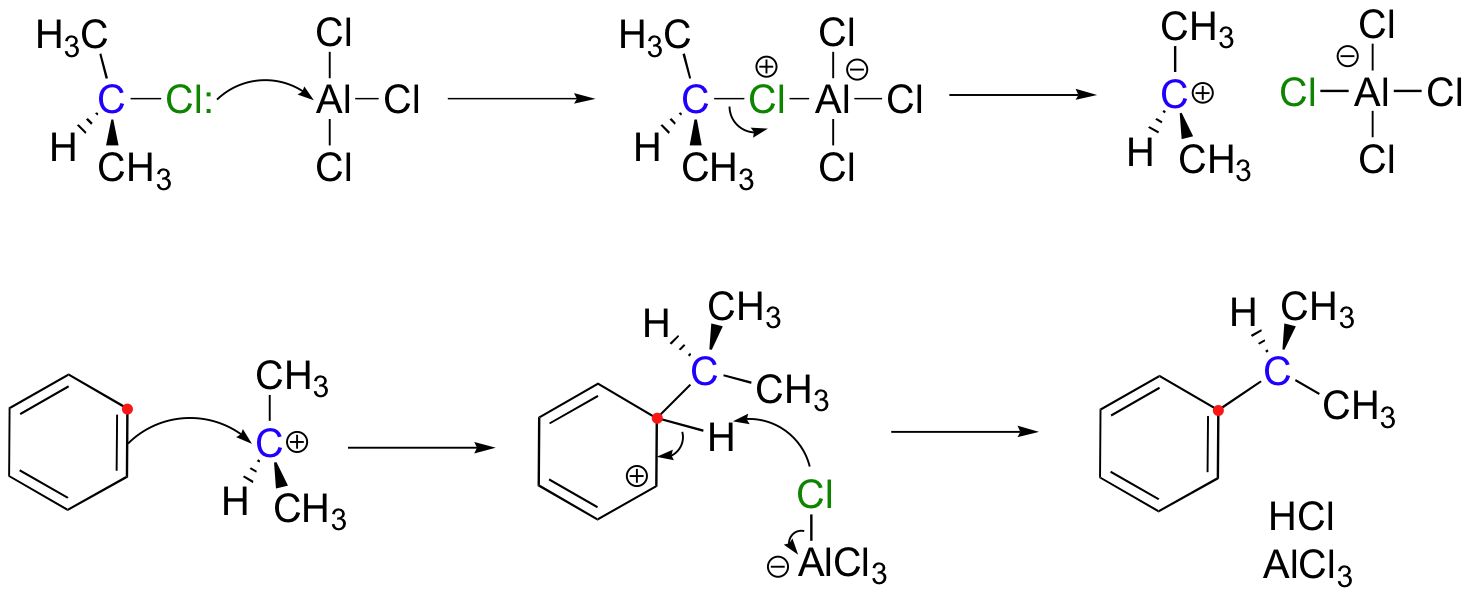
This SN1-like SEAr condensation is mechanistically analogous to the enzymatic tryptophan-DMAPP transferase reaction we saw in section 15.5B.
An alternate method for carrying out a Friedel-Crafts alkylation is to start with an alkene, which is protonated by a strong acid such as H2SO4 to generate a carbocation.

In Friedel-Frafts alkylation reactions, care must always be taken when choosing the alkylating compound because of the possibility of undesired carbocation rearrangements (this won’t make much sense right now, but it will after we cover carbocation rearrangements in section 15.7).
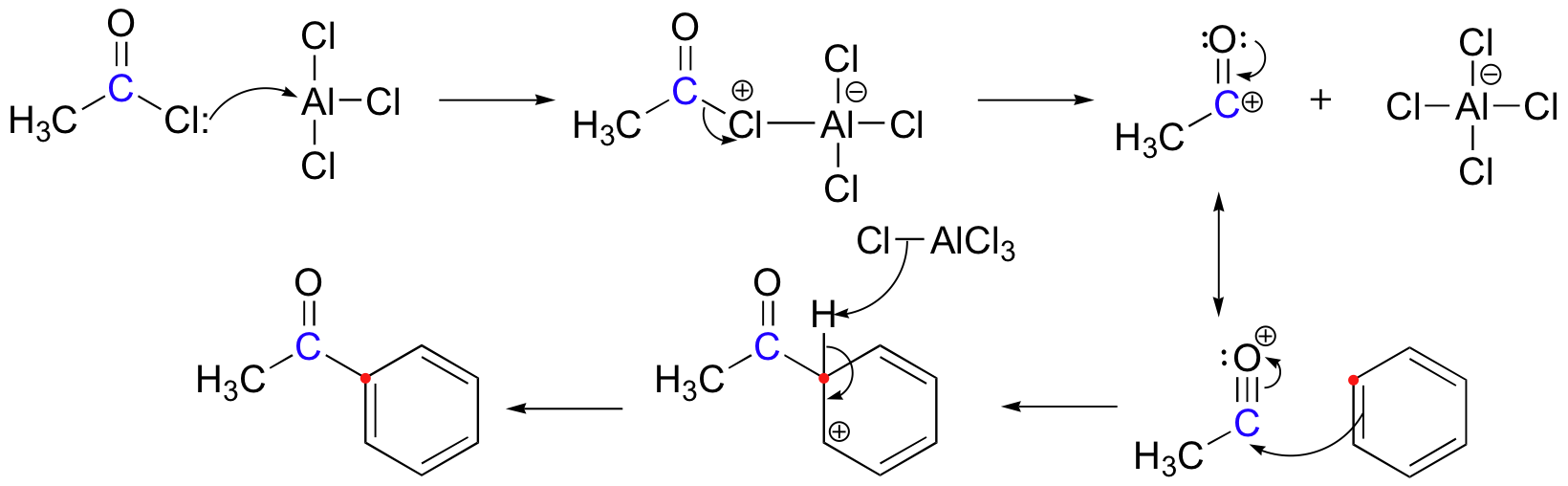
The Friedel-Crafts acylation reaction is essentially an acyl substitution (chapter 12) reaction with an aromatic π bond serving as the nucleophile. As in many other laboratory acyl transfer reactions, acyl chlorides (section 12.2D) are used as activated carboxylic acids. Because of the low reactivity of the aromatic π bond nucleophile, however, the acyl chloride electrophile in a Friedel-Crafts acylation must be further activated with a Lewis acid reagent such as aluminum trichloride, which again serves to polarize the carbon-chlorine bond and increase the electrophilicity of the acyl carbon. The activated electrophile in Friedel-Crafts acylations is often depicted as an ionic species.
Aromatic groups can be halogenated in electrophilic aromatic substitution reactions using molecular bromine or chlorine in combination with metal Lewis acid reagents such as AlCl3, FeCl3, or FeBr3.
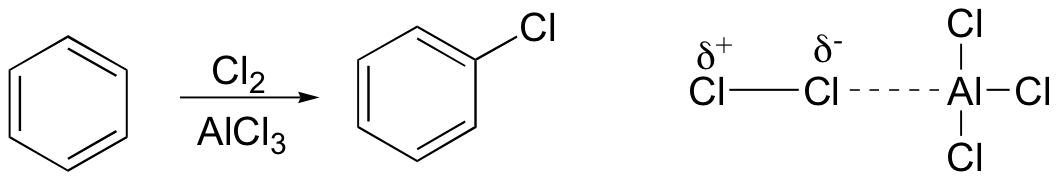
Just as in the Friedel crafts alkylation reaction the metal serves to polarize the halogen-halogen bond, thus creating a better electrophile.
Treating benzene with nitric acid and concentrated sulfuric acid yeilds nitrobenzene. The actual electrophile in the electrophilic aromatic substitution reaction is nitronium ion, NO2+.
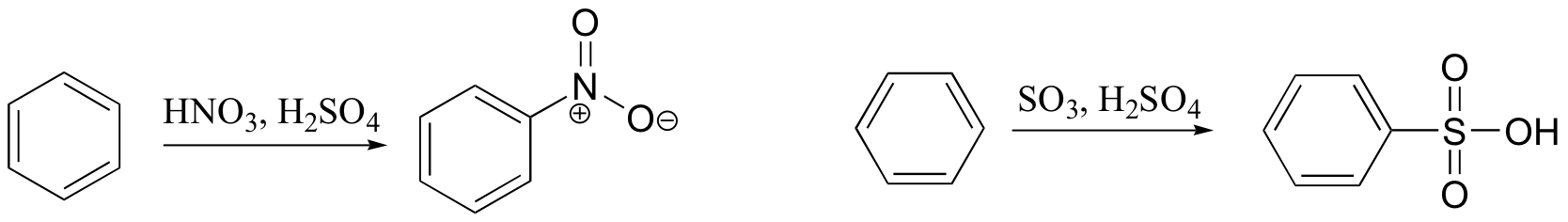
Sulfonation of benzene is accomplished by treatment with concentrated sulfuric acid containing a small amount of sulfur trioxide (referred to as fuming sulfuric acid). The actual electrophile in this case is sulfur trioxide.
15.6B: Ring-directing effects in Friedel-Crafts reactions
The reactivity of aromatic pi bonds in SEAr reactions is very sensitive to the presence of electron-donating groups (EDG) and electron-withdrawing groups (EWG) on the aromatic ring. This is due to the carbocation nature of the intermediate, which is stabilized by electron-donating groups and destabilized by electron-withdrawing groups.
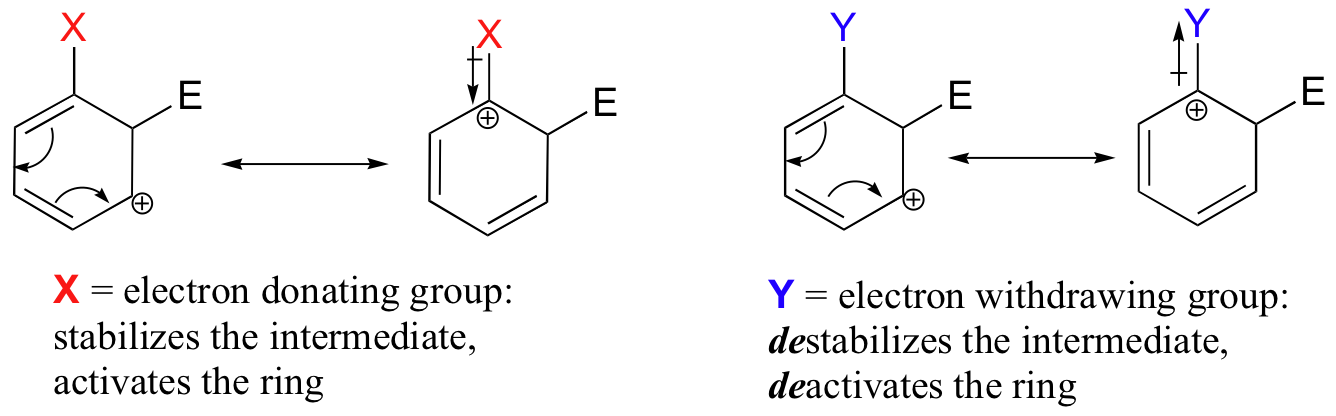
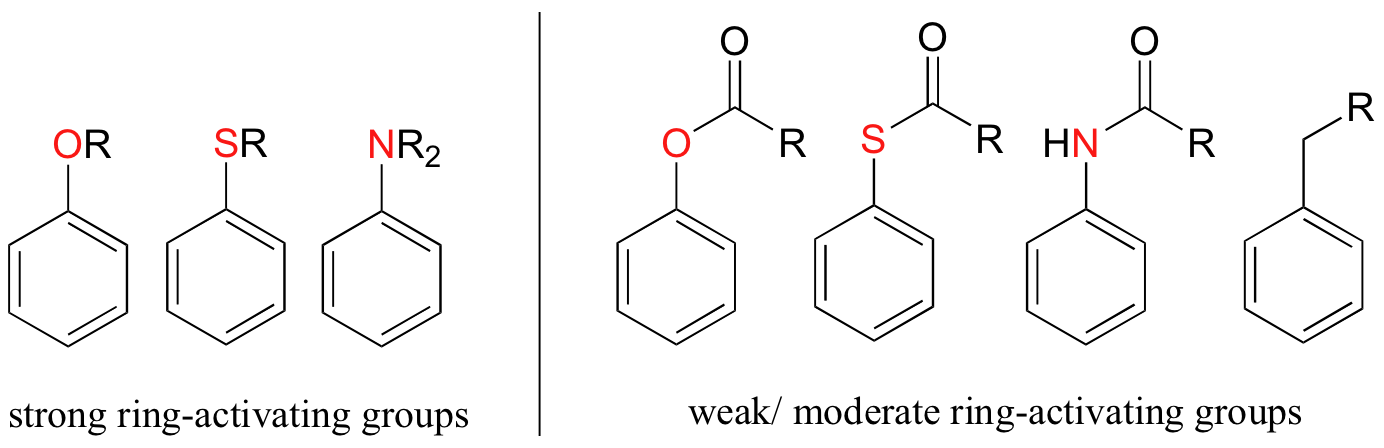
Alkyl groups are weakly ring-activating groups, as their electron-donating ability stems only from weak inductive effects. Substituents with heteroatoms connected to the aromatic ring are significantly more ring-activating than alkyl groups, because resonance electron-donating effects are possible. Amines, for example, are very powerful ring-activating substituents, due to the ability of the lone pair on the nitrogen to stabilize the carbocation intermediate through resonance:

Other ring-activating groups are shown below (in these figures, the R group can be a hydrogen). All of these groups are able, in varying degrees, to stabilize the carbocation intermediate in an electrophilic aromatic substitution reaction. Notice that plain old alkyl groups are also (weakly) ring-activating.
Substituent groups that are ring-activating due to resonance effects also tend to exert a strong regiochemical influence on further substitution reactions. Specifically, substitution tends to occur in the ortho and para positions relative to the existing group. This is known as the ortho-para directing effect. The effect can be explained by drawing resonance contributors for the carbocation intermediate of the SEAr reaction: the positive charge is in position to be delocalized by resonance only in reactions leading to ortho or para substitution.

The carbocation which leads to the meta-substituted product, however, cannot be stabilized by resonance with the ring-activating group:

As an example, the Friedel-Crafts alkylation of methoxy benzene would be expected to produce a mixture of the ortho and para substituted products, but no meta-substituted product.

In addition, the para product would be expected to be preferred over the ortho product, due to steric considerations.
Electron-withdrawing substituents on an aromatic ring are ring-deactivating, making it harder for further substitution reactions to occur. These are mostly carbonyl-containing groups, as well as alkyl halides.
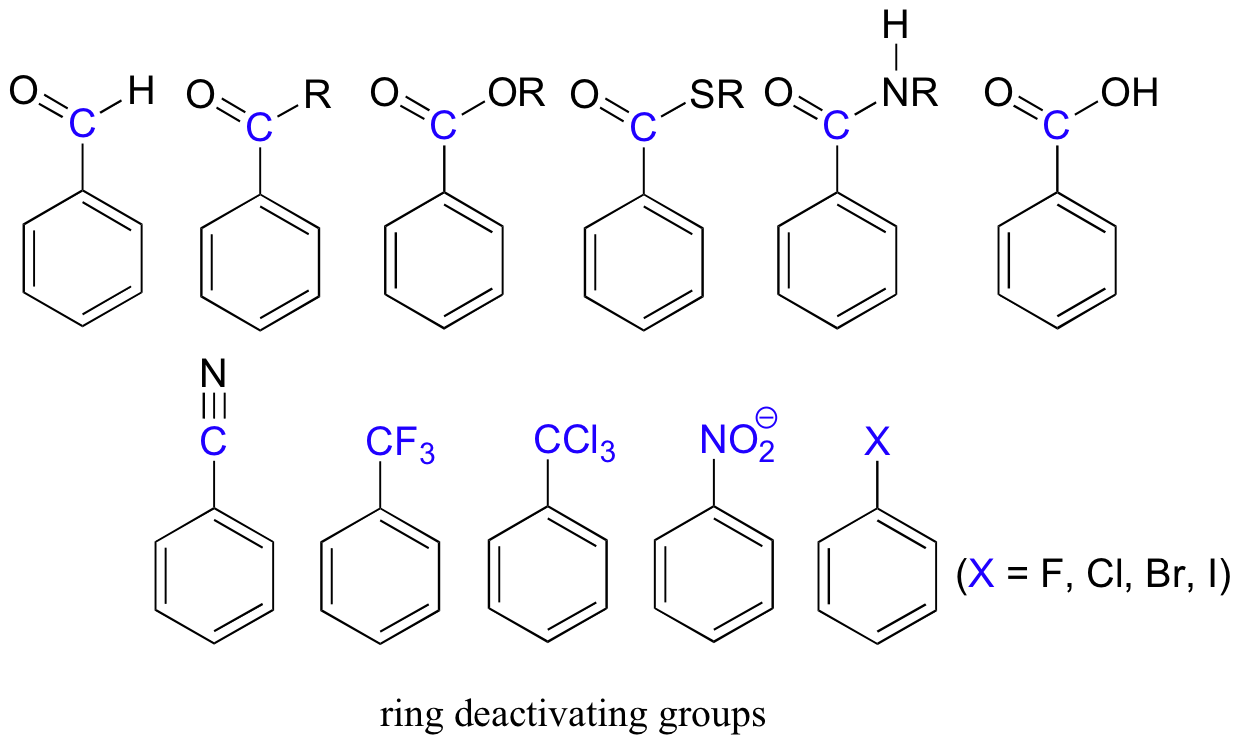
When substitution does occur on an aromatic ring with deactivating group already attached, it tends to occur specifically at the meta position - deactivating groups are generally meta-directing. The exception to this rule is the halides, which are ring-deactivating but ortho-para directing. You will get a chance to design some syntheses involving Friedel-Craft reactions and associated ring-directing effects in the end-of-chapter problems.
As a final note, if you look back at section 15.5B, you will notice that all of the enzymatic SEAr reactions discussed there took place on aromatic rings with heteroatom-containing ring-activating groups, and that the positive charges on the reaction intermediates were always positioned so that they could be delocalized to the heteroatom. Nature likes to make things easy whenever possible!