
Chapter 9 Solutions
- Page ID
- 1124

In-chapter exercises
E9.1: The electrophilic carbon is primary, and a carbocation intermediate would not be stabilized by resonance. Therefore it is reasonable to predict an SN2 mechanism.
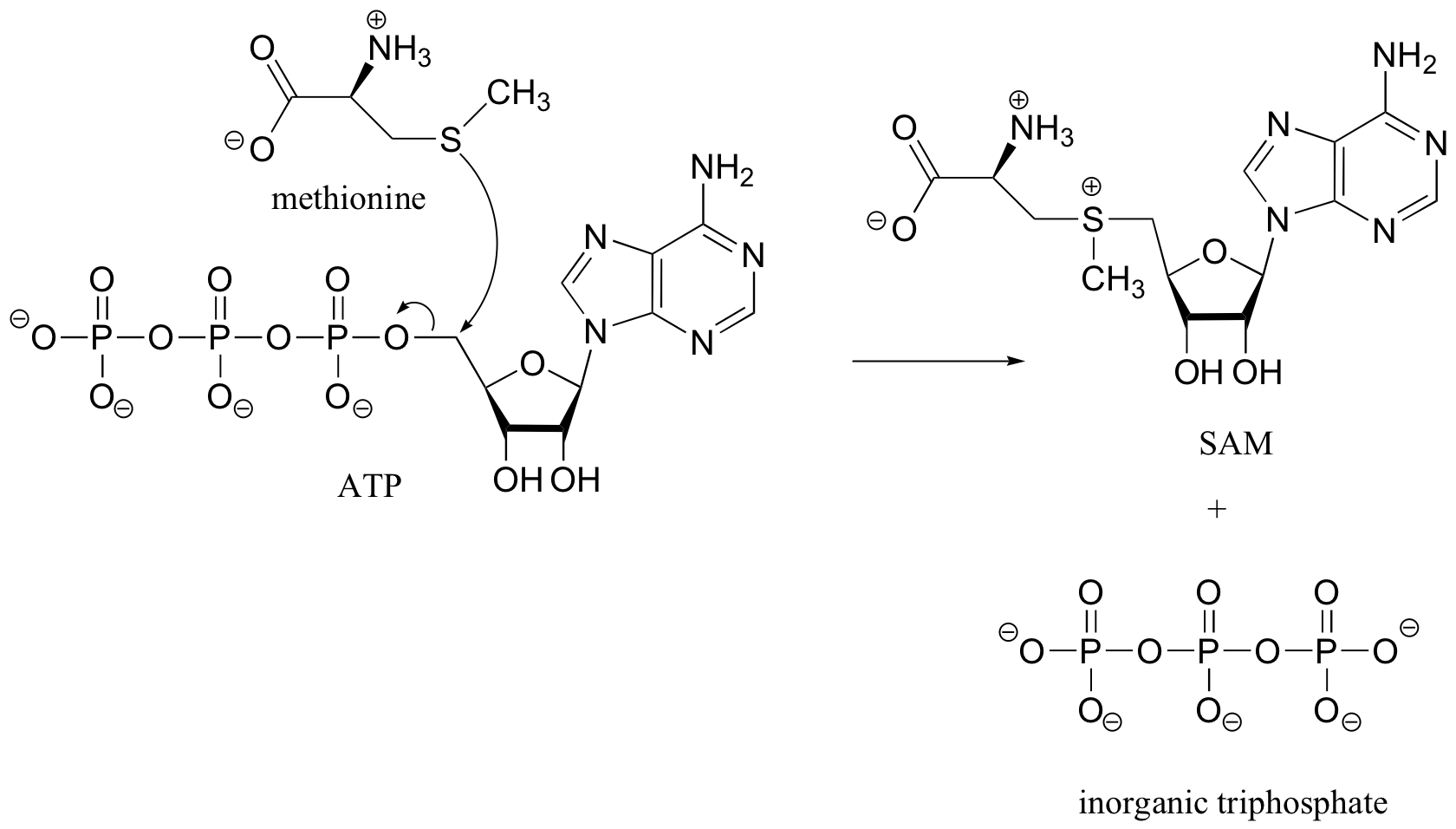
Note: in later chapters, especially in chapters 10 and 12, we will see many more reactions involving ATP. As we will see then, the SAM-forming reaction above is somewhat unusual – most reactions with ATP involve nucleophilic attack at one of the phosphate groups, rather than at the methylene (CH2) carbon.
E9.2: Due to the relatively high nucleophilicity of the amine group, it does not need to be deprotonated prior to attack. The deprotonation step takes place after the nucleophilic attack, when an ammonium ion is the proton donor (an ammonium ion is a relative strong acid and does not require a strong base for deprotonation). The alcohol, in contrast, is less nucleophilic and needs to be deprotonated by a strong base before acting as a nucleophile in an SN2 reaction.
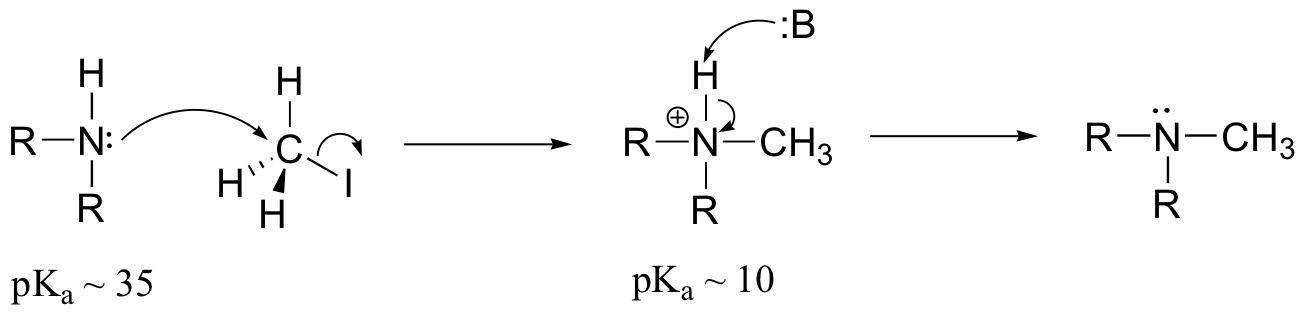
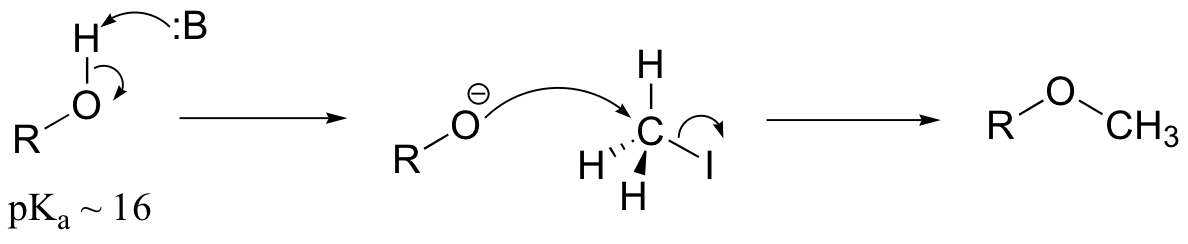
E9.3: The nucleophile in this reaction is an amine, as opposed to a thiolate in the protein farnesyltransferase reaction. A thiolate is a very powerful nucleophile, an amine somewhat less so. Therefore the N-alkylating reaction, with the weaker nucleophile, is likely to be more SN1 in character, and thus would be expected to exhibit higher sensitivity to fluorine substitution on the electrophilic substrate (in other words, more positive charge develops on C1 of the electrophile in the AMP reaction)
E9.4: The SN1 model is a solvolysis reaction (water is the nucleophile), which as a rule proceed through carbocation intermediates. In the SN2 model the nucleophile is a relatively powerful iodide ion, a factor which favors a concerted mechanism.
E9.5: Notice that in the second step, when water enters the picture, stereochemistry is conserved. If the step were indeed a direct nucleophilic displacement by water, the observed stereochemistry would be different: either inversion as show below in the case of an SN2 mechanism, or racemization in the case of an SN1 mechanism.
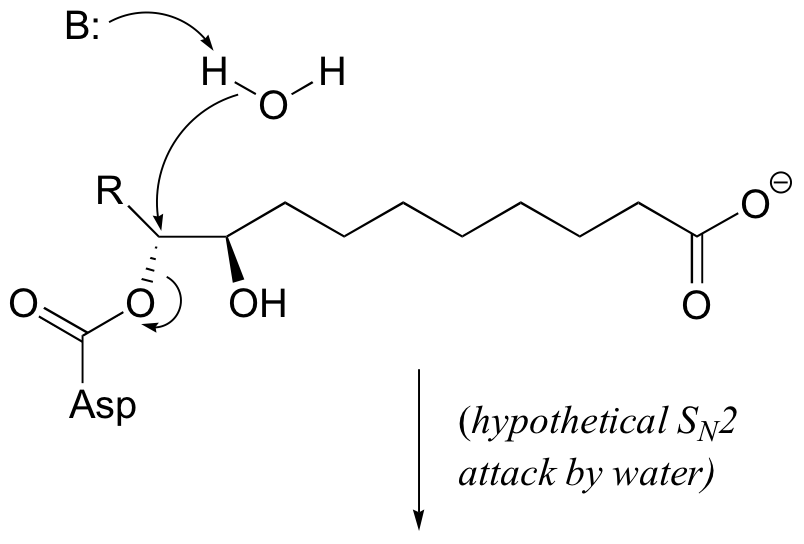
E9.6: This is essentially a nucleophilic (or ‘base-catalyzed’) ring-opening, with the nucleophilic thiol attacking the less hindered carbon of the epoxide. The right-side epoxide carbon is ‘protected’ from nucleophilic attack by the negatively charged oxygens of the phosphonate group.
End-of-chapter problems
P9.1:
a)
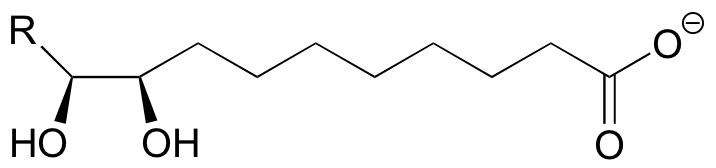
P9.2: (See Biochem 42, 1564)
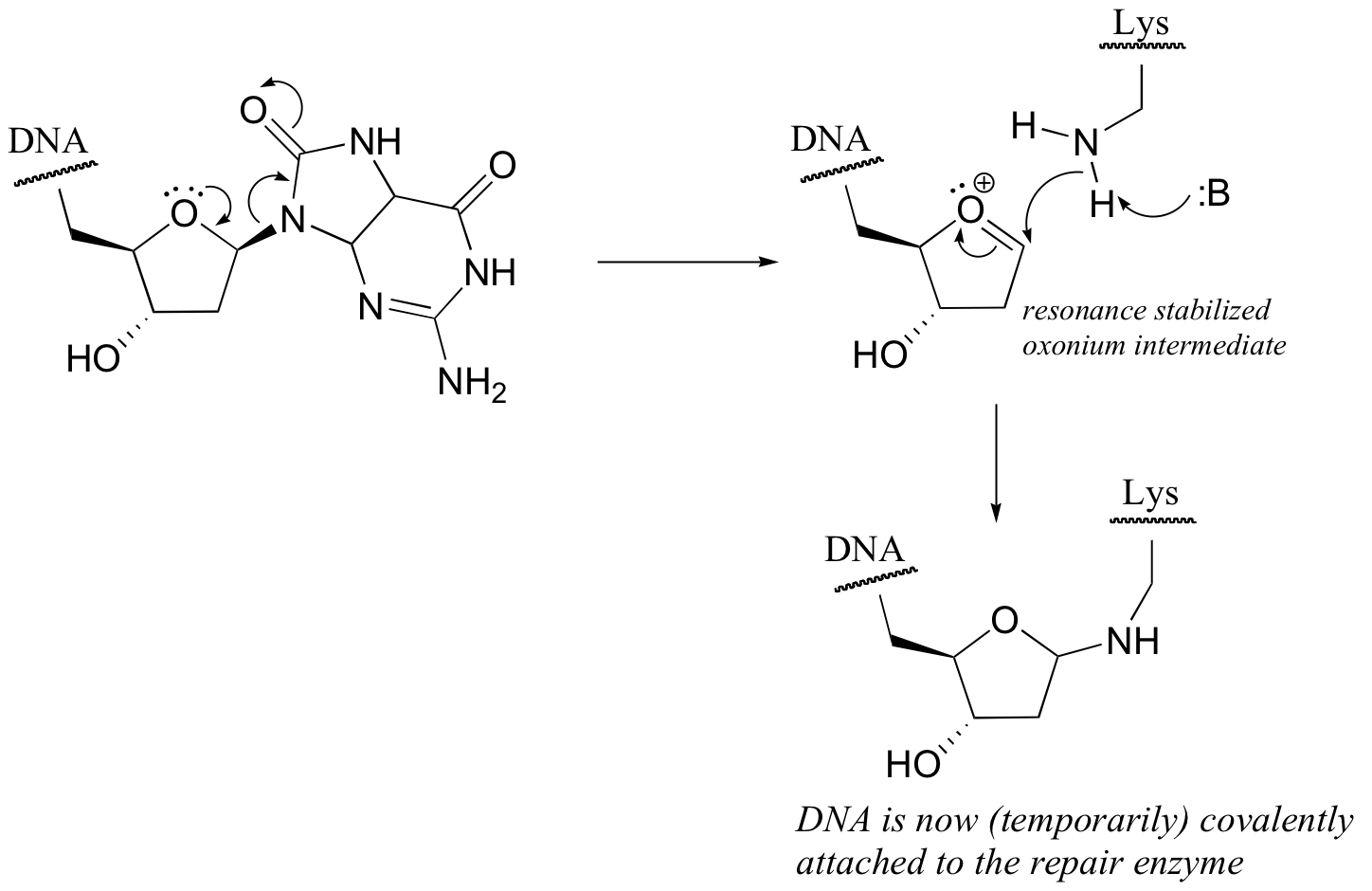
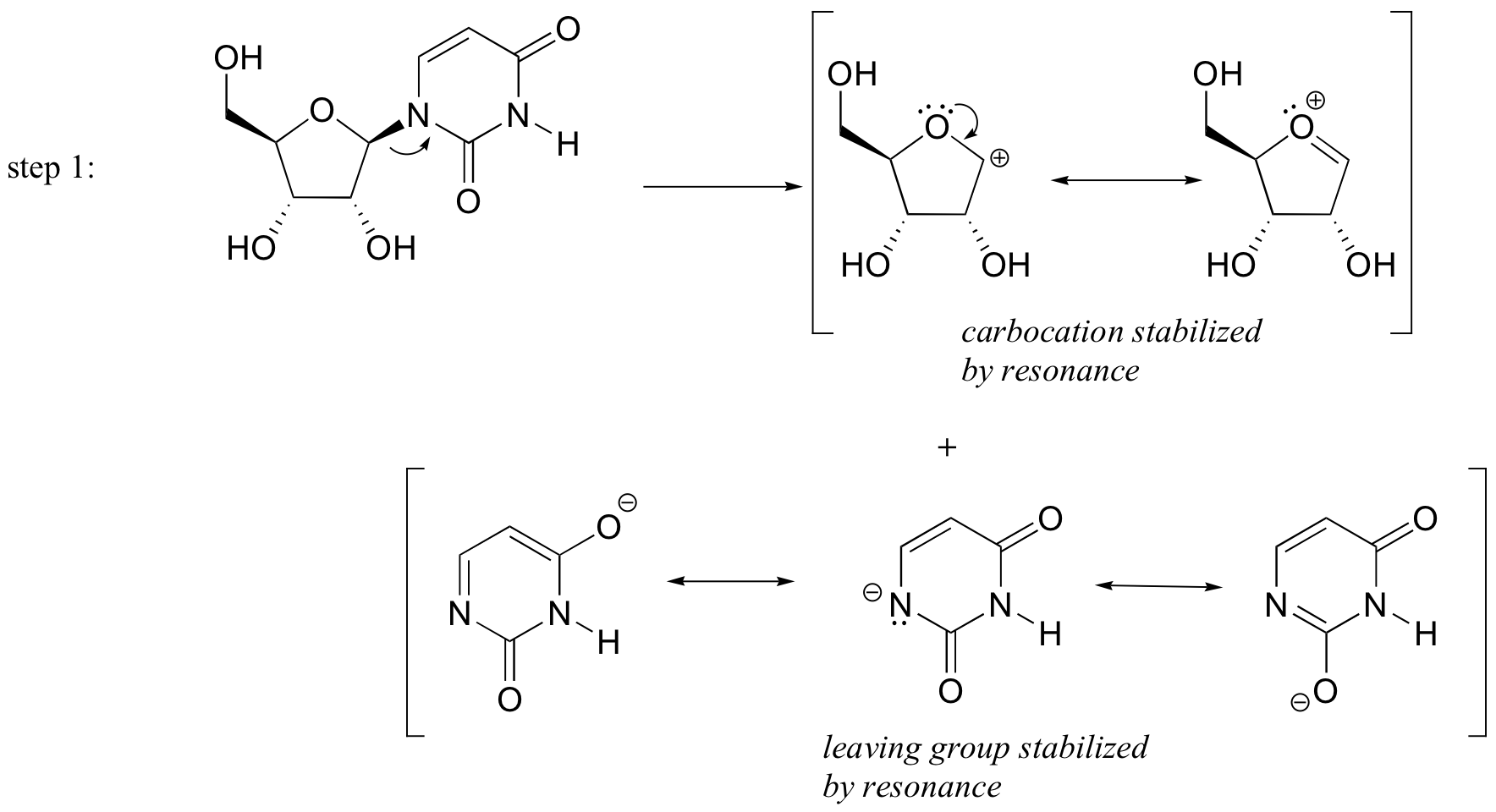
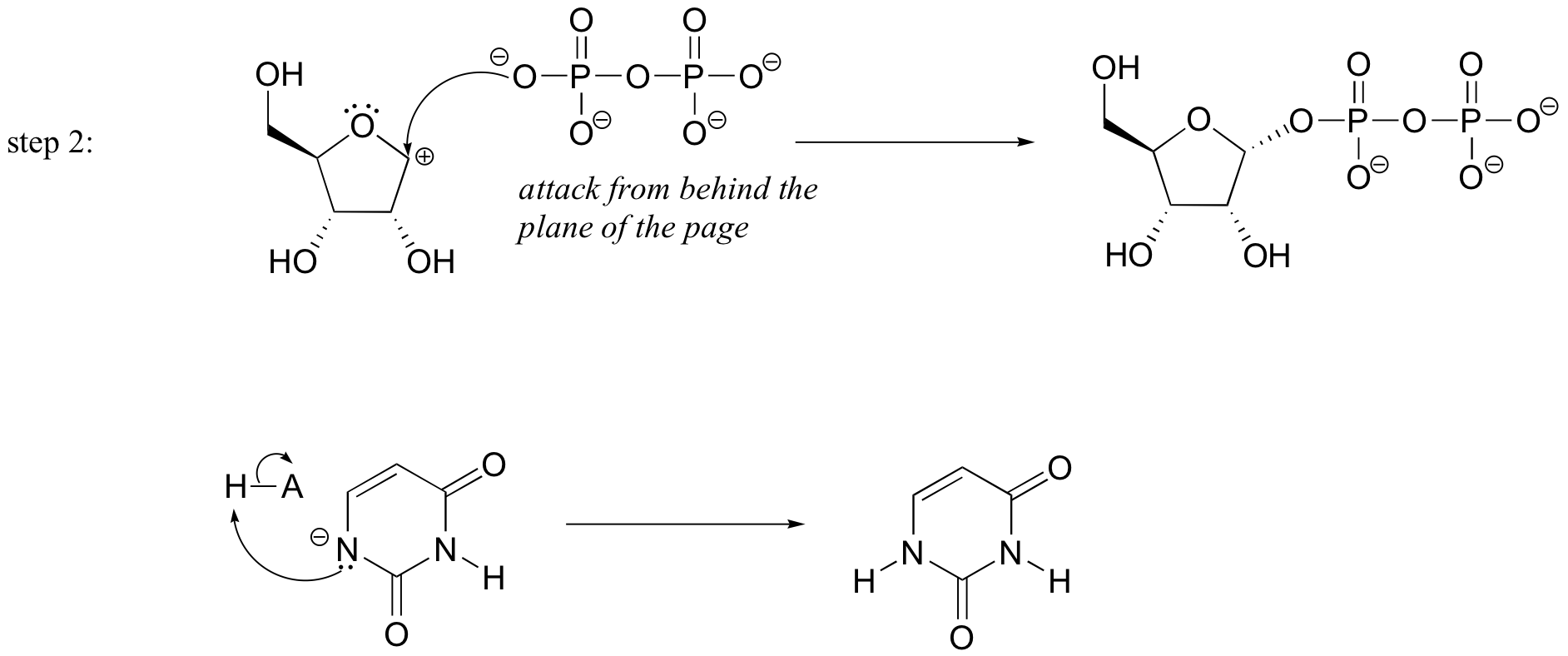
P9.3:
a) Because the reaaction involves the transfer of a methyl group to an amine, the most likely biomolecule would be S-adenosylmethionine (SAM – see section 9.1).
b) A mechanism with an abbreviated version of the SAM structure is shown below. This is an SN2 mechanism, similar to other SAM-dependent methyltransferase reactions.
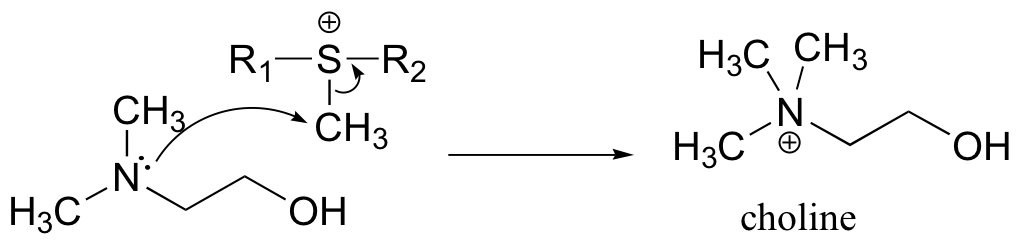
P9.4:
a) Because the process involves two SN2 displacements, it is reasonable to predict that it is the thiol group – the most nucleophilic group in glutathione – that is playing the key role. For this reason, we will use the abbreviation GSH for glutathione in the mechanism in part b) below.
b)
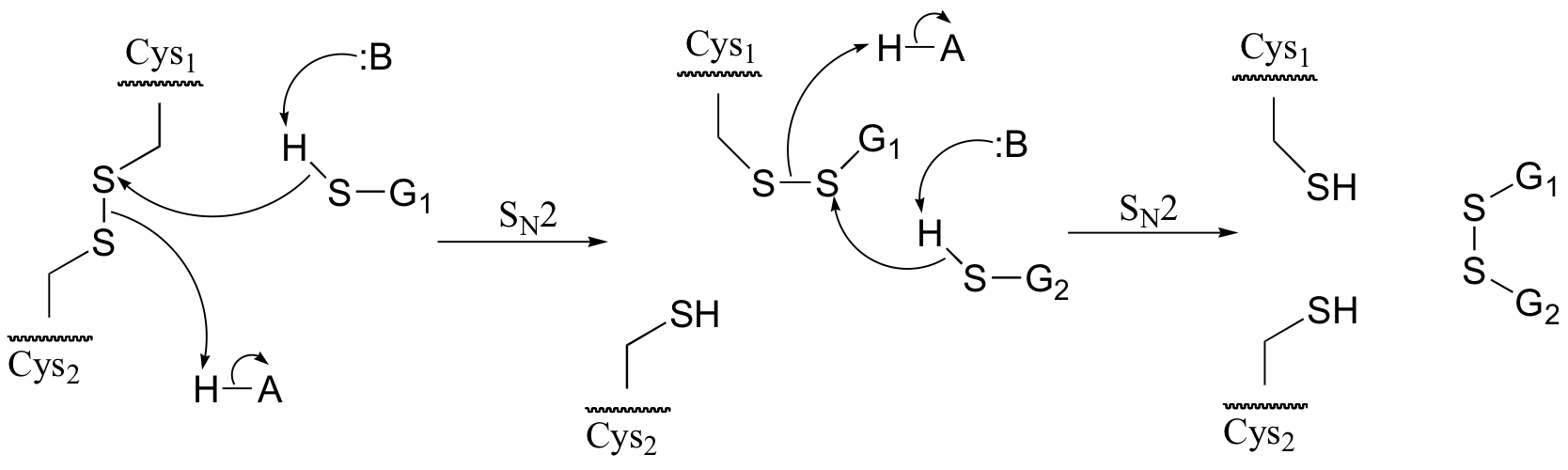
P9.5: (See Acc Chem. Res. 2001, 34, 681.) A likely mechanism could involve two successive SN2 methyl transfer steps, the first from 5-methyltetrahydrofolate to cobalamin, and the second from cobalamin to homocysteine, the direct precursor to methionine. This would account for the observed retention of configuration.
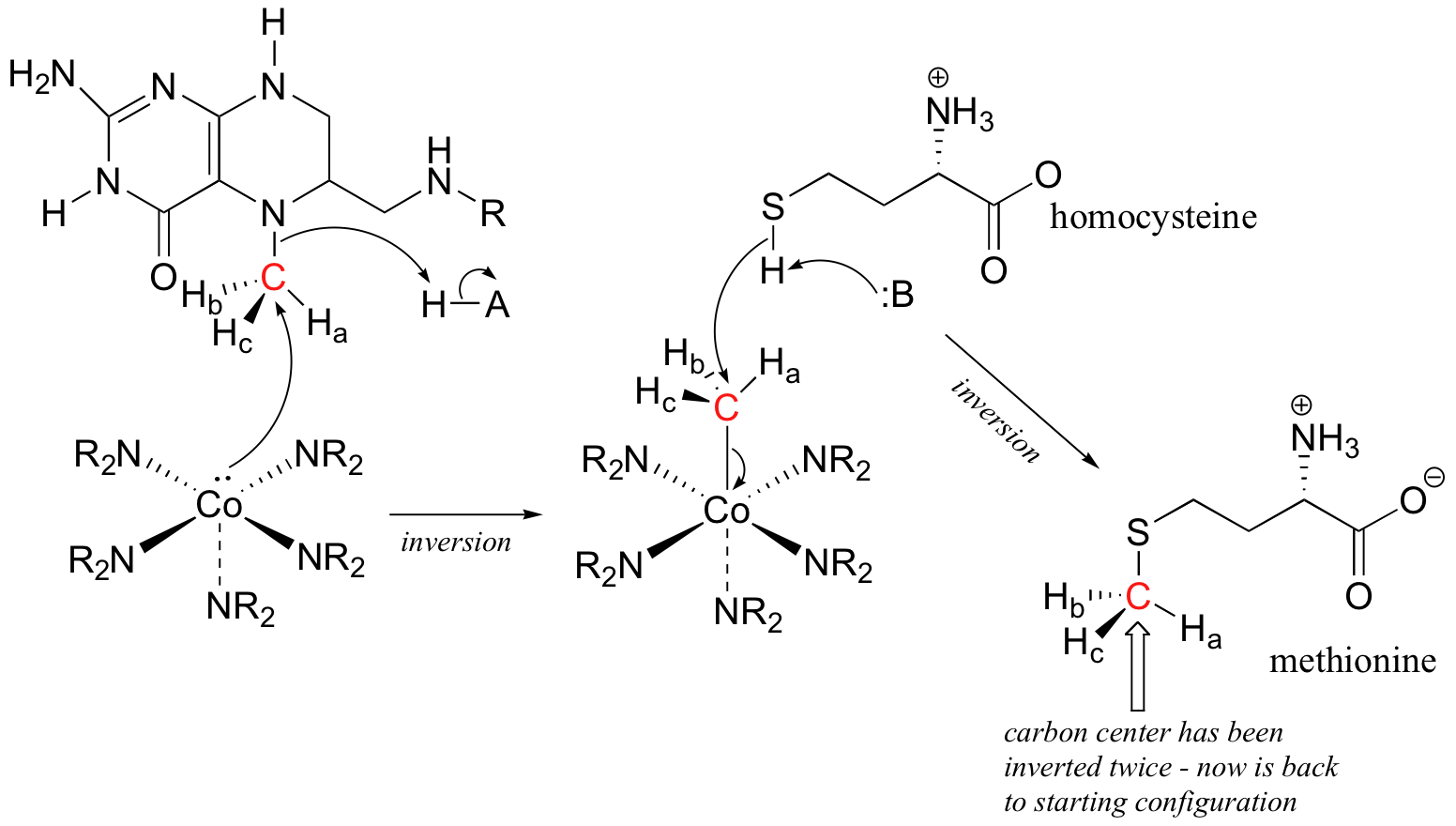
P9.6:
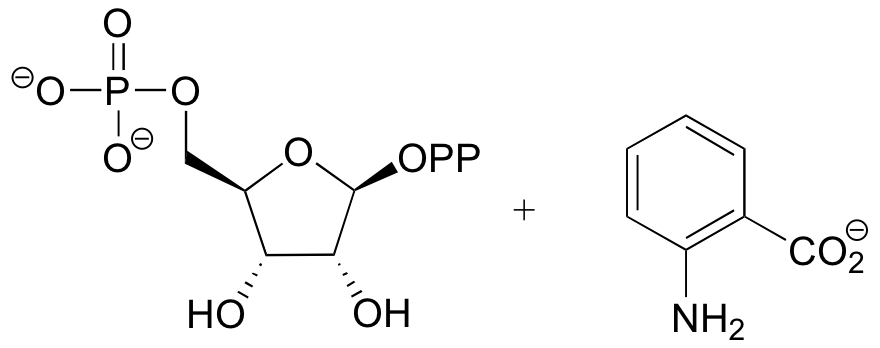
Challenge problems
C9.1: See Biochem 2005, 44, 8989; Structure 2004, 12, 927.
C9.2: See Biochem 1983, 22, 806; Chem Res Toxicol 1997, 10, 2 (scheme 1).
C9.3: See Chem Res Toxicol 1997, 10, 2 (fig 13)
C9.4: See Biochem 43, 7187 (look at compound #6 just above the Materials and Methods section):
C9.5:
a,b) See J. Am. Chem. Soc. 1994, 116, 11594.
c) See J. Am. Chem. Soc. 1987, 109, 7530.
Contributors
- Organic Chemistry With a Biological Emphasis, Tim Soderberg (University of Minnesota, Morris)