
Carbocation Rearrangements
- Page ID
- 1136
Carbocation rearrangements are extremely common in organic chemistry reactions are are defined as the movement of a carbocation from an unstable state to a more stable state through the use of various structural reorganizational "shifts" within the molecule. Once the carbocation has shifted over to a different carbon, we can say that there is a structural isomer of the initial molecule. However, this phenomenon is not as simple as it sounds.
Introduction
Whenever alcohols are subject to transformation into various carbocations, the carbocations are subject to a phenomenon known as carbocation rearrangement. A carbocation, in brief, holds the positive charge in the molecule that is attached to three other groups and bears a sextet rather than an octet. However, we do see carbocation rearrangements in reactions that do not contain alcohol as well. Those, on the other hand, require more difficult explanations than the two listed below. There are two types of rearrangements: hydride shift and alkyl shift. These rearrangements usualy occur in many types of carbocations. Once rearranged, the molecules can also undergo further unimolecular substitution (SN1) or unimolecular elimination (E1). Though, most of the time we see either a simple or complex mixture of products. We can expect two products before undergoing carbocation rearrangement, but once undergoing this phenomenon, we see the major product.
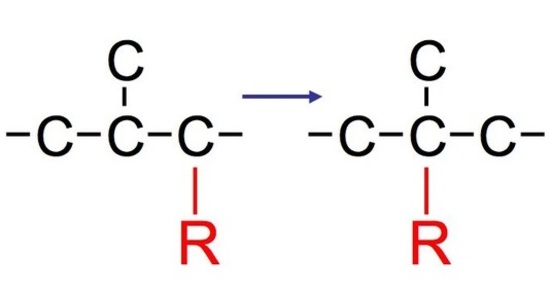
Hydride Shift
Whenever a nucleophile attacks some molecules, we typically see two products. However, in most cases, we normally see both a major product and a minor product. The major product is typically the rearranged product that is more substituted (aka more stable). The minor product, in contract, is typically the normal product that is less substituted (aka less stable).
The reaction: We see that the formed carbocations can undergo rearrangements called hydride shift. This means that the two electron hydrogen from the unimolecular substitution moves over to the neighboring carbon. We see the phenomenon of hydride shift typically with the reaction of an alcohol and hydrogen halides, which include HBr, HCl, and HI. HF is typically not used because of its instability and its fast reactivity rate. Below is an example of a reaction between an alcohol and hydrogen chloride:
.jpg?revision=1&size=bestfit&width=410&height=306)
GREEN (Cl) = nucleophile BLUE (OH) = leaving group ORANGE (H) = hydride shift proton RED(H) = remaining proton
The alcohol portion (-OH) has been substituted with the nucleophilic Cl atom. However, it is not a direct substitution of the OH atom as seen in SN2 reactions. In this SN1 reaction, we see that the leaving group, -OH, forms a carbocation on Carbon #3 after receiving a proton from the nucleophile to produce an alkyloxonium ion. Before the Cl atom attacks, the hydrogen atom attached to the Carbon atom directly adjacent to the original Carbon (preferably the more stable Carbon), Carbon #2, can undergo hydride shift. The hydrogen and the carbocation formally switch positions. The Cl atom can now attack the carbocation, in which it forms the more stable structure because of hyperconjugation. The carbocation, in this case, is most stable because it attaches to the tertiary carbon (being attached to 3 different carbons). However, we can still see small amounts of the minor, unstable product. The mechanism for hydride shift occurs in multiple steps that includes various intermediates and transition states. Below is the mechanism for the given reaction above:
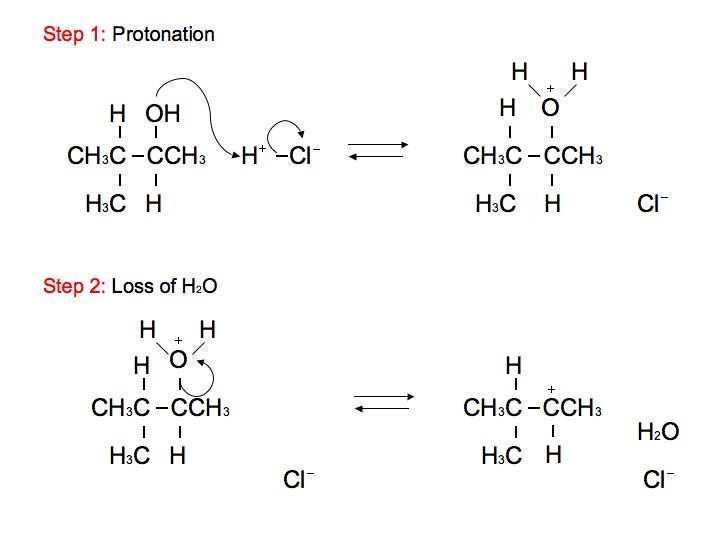
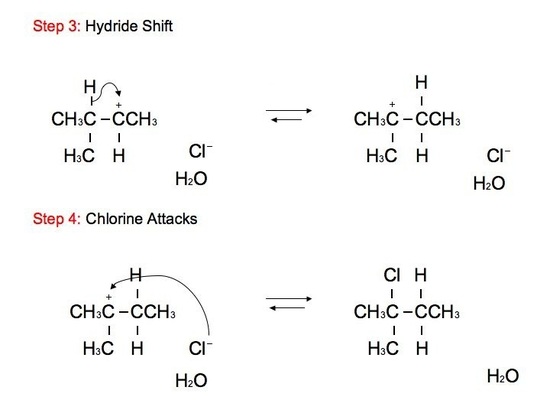
Hydration of Alkenes: Hydride Shift
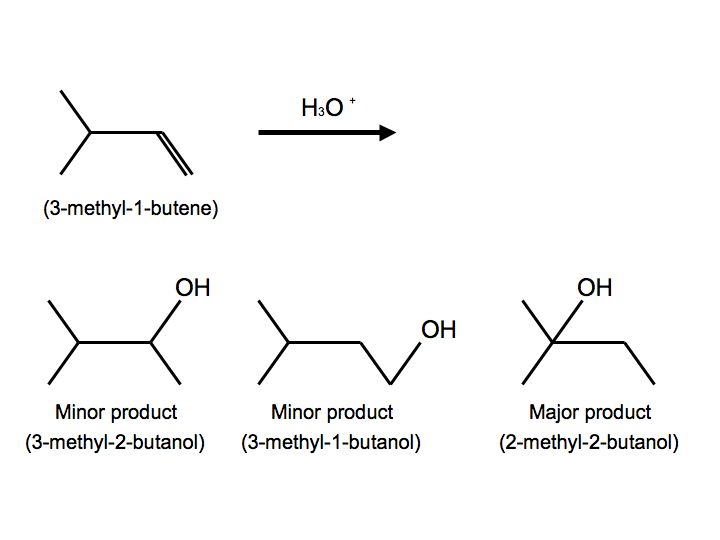
In a more complex case, when alkenes undergo hydration, we also observe hydride shift. Below is the reaction of 3-methyl-1-butene with H3O+ that furnishes to make 2-methyl-2-butanol:
.jpg?revision=1&size=bestfit&width=456&height=340)
Once again, we see multiple products. In this case, however, we see two minor products and one major product. We observe the major product because the -OH substitutent is attached to the more substituted carbon. When the reactant undergoes hydration, the proton attaches to carbon #2. The carbocation is therefore on carbon #2. Hydride shift now occurs when the hydrogen on the adjacent carbon formally switch places with the carbocation. The carbocation is now ready to be attacked by H2O to furnish an alkyloxonium ion because of stability and hyperconjugation. The final step can be observed by another water molecule attacking the proton on the alkyloxonium ion to furnish an alcohol. We see this mechanism below:
.jpg?revision=1&size=bestfit&width=512&height=383)
Alkyl Shift
Not all carbocations have suitable hydrogen atoms (either secondary or tertiary) that are on adjacent carbon atoms available for rearrangement. In this case, the reaction can undergo a different mode of rearrangement known as alkyl shift (or alkyl group migration). Alkyl Shift acts very similarily to that of hydride shift. Instead of the proton (H) that shifts with the nucleophile, we see an alkyl group that shifts with the nucleophile instead. The shifting group carries its electron pair with it to furnish a bond to the neighboring or adjacent carbocation. The shifted alkyl group and the positive charge of the carbocation switch positions on the moleculeReactions of tertiary carbocations react much faster than that of secondary carbocations. We see alkyl shift from a secondary carbocation to tertiary carbocation in SN1 reactions:
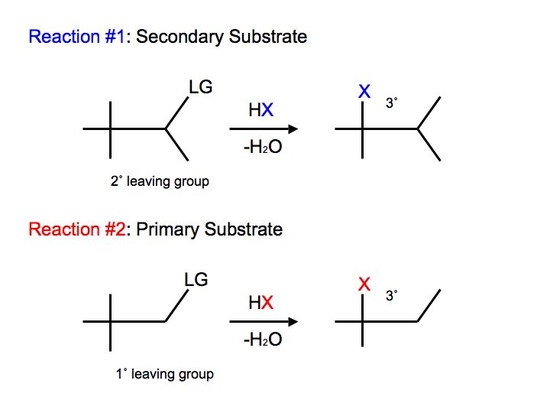
We observe slight variations and differences between the two reactions. In reaction #1, we see that we have a secondary substrate. This undergoes alkyl shift because it does not have a suitable hydrogen on the adjacent carbon. Once again, the reaction is similar to hydride shift. The only difference is that we shift an alkyl group rather than shift a proton, while still undergoing various intermediate steps to furnish its final product.
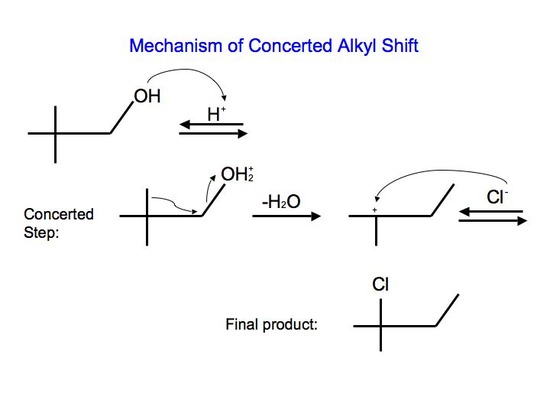
With reaction #2, on the other hand, we can say that it undergoes a concerted mechanism. In short, this means that everything happens in one step. This is because primary carbocations cannot be an intermediate and they are relatively difficult processes since they require higher temperatures and longer reaction times. After protonating the alcohol substrate to form the alkyloxonium ion, the water must leave at the same time as the alkyl group shifts from the adjacent carbon to skip the formation of the unstable primary carbocation.
Carbocation Rearrangements for E1 Reactions
E1 reactions are also affected by alkyl shift. Once again, we can see both minor and major products. However, we see that the more substituted carbons undergo the effects of E1 reactions and furnish a double bond. See practice problem #4 below for an example as the properties and effects of carbocation rearrangements in E1 reactions are similar to that of alkyl shifts.
1,3-Hydride and Greater Shifts
Typically, hydride shifts can occur at low temperatures. However, by heating the solutionf of a cation, it can easily and readily speed the process of rearrangement. One way to account for a slight barrier is to propose a 1,3-hydride shift interchanging the functionality of two different kinds of methyls. Another possibility is 1,2 hydride shift in which you could yield a secondary carbocation intermediate. Then, a further 1,2 hydride shift would give the more stable rearranged tertiary cation.
More distant hydride shifts have been observed, such as 1,4 and 1,5 hydride shifts, but these arrangements are too fast to undergo secondary cation intermediates.
Analogy
Carbocation rearrangements happen very readily and often occur in many organic chemistry reactions. Yet, we typically neglect this step. Dr. Sarah Lievens, a Chemistry professor at the University of California, Davis once said carbocation rearrangements can be observed with various analogies to help her students remember this phenomenon. For hydride shifts: "The new friend (nucleophile) just joined a group (the organic molecule). Because he is new, he only made two new friends. However, the popular kid (the hydrogen) glady gave up his friends to the new friend so that he could have even more friends. Therefore, everyone won't be as lonely and we can all be friends." This analogy works for alkyl shifts in conjunction with hydride shift as well.
References
- Vogel, Pierre. Carbocation Chemistry. Amsterdam: Elsevier Science Publishers B.V., 1985.
- Olah, George A. and Prakash, G.K. Surya. Carbocation Chemistry. New Jersey: John Wiley & Sons, Inc., 2004.
- Vollhardt, K. Peter C. and Schore, Neil E. Organic Chemistry: Structure and Function. New York: Bleyer, Brennan, 2007.
Outside links
Problems
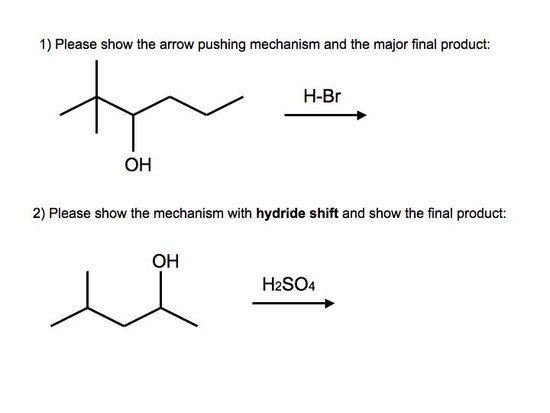
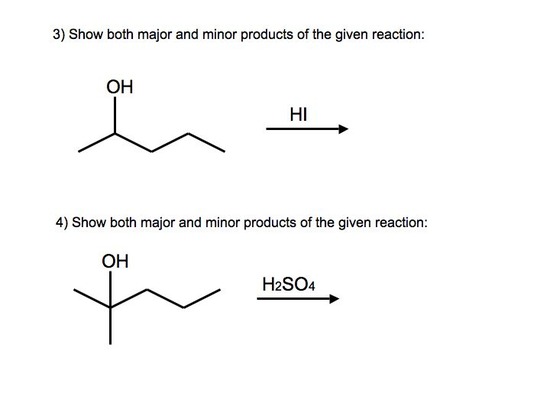
Answers to Practice Problems
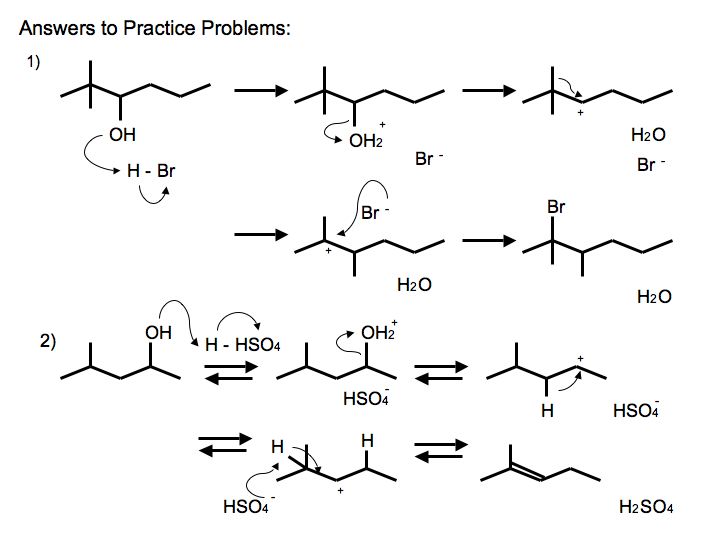
.jpg?revision=2&size=bestfit&width=585&height=439)
Contributors
- Jeffrey Ma