
Chemiluminescence
- Page ID
- 1760
Most chemiluminescence methods involve only a few chemical components to actually generate light. Luminol chemiluminescence (Nieman, 1989), which has been extensively investigated, and peroxyoxalate chemiluminescence (Given and Schowen, 1989; Orosz et al., 1996) are both used in bioanalytical methods and will be the subject of this primer on chemiluminescence. In each system, a "fuel" is chemically oxidized to produced an excited state product. In many luminol methods it is this excited product that emits the light for the signal. In peroxyoxalate chemiluminescence, the initial excited state product does not emit light at all and instead it reacts with another compound.
Introduction
Chemiluminescence takes its place among other spectroscopic techniques because of its inherent sensitivity and selectivity. It requires:
- no excitation source (as does fluorescence and phosphorescence)
- only a single light detector such as a photomultiplier tube
- no monochromator and often not even a filter
Maybe this list should be entitled "What chemiluminescent systems do not require." Although not as widely applicable as excitation spectroscopy, the detection limits for chemiluminescent methods can be 10 to 100 times lower than other luminescence techniques.
Most chemiluminescence methods involve only a few chemical components to actually generate light. Luminol chemiluminescence (Nieman, 1989), which has been extensively investigated, and peroxyoxalate chemiluminescence (Given and Schowen, 1989; Orosz et al., 1996) are both used in bioanalytical methods and will be the subject of this primer on chemiluminescence. In each system, a "fuel" is chemically oxidized to produced an excited state product. In many luminol methods it is this excited product that emits the light for the signal. In peroxyoxalate chemiluminescence, the initial excited state product does not emit light at all and instead it reacts with another compound, often a compound also viable as a fluorescent dye, and it is this fluorophore which becomes excited and emits light. That said, the oxalate reactions, to have practical applicability in, for instance HPLC, require a mixed solvent system (buffer/organic solvent) to assure solubility of the reagents, optimized pH, and allow compatibility with the analytes.
A general discussion of these two methods, their applicability as reported in some of the recent literature, and a discussion of the emission spectra of each--complete with movies that show short experiments with each--will be presented.
Peroxyoxalate Chemiluminescence Primer
One of the suggested reaction sequences in the reaction of peroxyoxalates, of which bis(2,4,6-trichlorophenyl)oxlate (TCPO) is the most prominent example, follows. It involves the fuel (TCPO) plus the oxidant (H2O2) reacting to produce a proposed intermediate, in this example shown as a dioxetane; although, this reaction probably produces many intermediates, and others, such as hydroperoxyoxalate, have been proposed (Milofsky and Birks, 1991; Choksi et al., 1990).
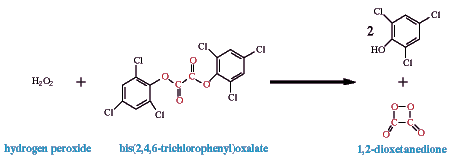
The intermediate, shown here as 1,2-dioxetanedione, excites a fluorophore. In the included movie demonstrating TCPO chemiluminescence, 9,10-diphenylanthracene acts as the fluorophore; its lambda max is 425 nm in the solvent used, tetrahydrofuran. Its reaction with the intermediate produces the excited state product which quickly emits light.
The process of transferring the energy of the initial reaction, the chemical reaction of hydrogen peroxide with TCPO, to light emission from the excited state fluorophore (fluorophore*) can be sidetracked along the way by loses in each step of the process: the initial oxidation to produce the intermediate, the reaction of the intermediate with a fluorophore, and the reaction of the excited fluorophore to produce light (Orosz et al., 1996).
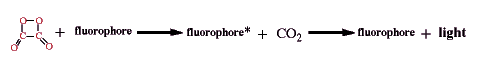
The initial oxidation can yield the high energy intermediate or
- TCPO can be hydrolyzed instead or
- oxidation can occur that doesn't yield chemiluminescent products.
The high energy intermediate can react to excite the fluorophore or
- the intermediate can react with a quencher more easily oxidized than the fluorophore
- the intermediate and fluorophore can react without yielding excited fluorophore
- the intermediate can decompose or be further oxidized by residual H2O2.
Finally the excited fluorophore can loose energy by emission of light or
- the excited fluorophore can de-excited by production of heat instead of light.
In normal chromatographic (HPLC) procedures, these alternate mechanistic routes can be effected by solvent and buffers (Orosz, 1989; Jennings and Capomacchia, 1988); pH (de Jong et al., 1986); catalyst (Orlovic et al., 1989; Alverez et al., 1986); and type of fuel (Orlovic et al., 1989; Orosz, 1989), oxidant (Orlovic et al., 1989), and fluorophore concentration and identify. Possibly most important for chromatographers, eluent and reagent flows (Givens and Schowen, 1989; Kwakman and Brinkman, 1992), detector volume and geometry (de Jong et al., 1990; Grayeski and Weber, 1984), and mixing parameters (Kobayashi and Imai, 1980; Sugiura et al., 1993) can all effect this method's light production.
This, therefore, sets the stage for analytical methods whereby manipulating the appropriate parameter allows for the sensitive determination of hydrogen peroxide (Pontén et al., 1996; Stigbrand et al., 1994) or fluorophore content.
Recently, for example, Hamachi et al. (1999) determined the concentration of propentofylline in hypocampus extracts from rats by derivitizing the analyte to create a fluorophore which would chemiluminesce with another peroxyoxalate, TDPO [bis(2-(3,6,9-trioadecanyloxycarbonyl)-4-nitrophenyl)oxalate, and hydrogen peroxide following HPLC. Propentofylline is a reported inhibitor of dopamine released during low oxygenation events in the cerebellum. The derivatization of propentofylline was carried out in trifluoracetic acid/acetonitrile solution using DBD-H (a benzoaxadiazole). The detection limit for the analyte, 31 fg/injection, was about 200 times better than comparable HPLC-UV methods.
Emission Spectrum of Diphenylanthracene as Chemiluminescent Fluorophore
A solution of TCPO and 9,10-diphenylanthracene (DPA; Aldrich Chemicals Co., Milwaukee, WI USA) both in the 1 x 10-3 M concentration range dissolved in tetrahydrofuran (THF) were mixed with a dilute solution of H2O2 in THF (~0.3%) at ~25oC. The resulting emission spectrum was recorded on a fluorescence spectrometer (Hitachi F-4500; 1 cm quartz cell) in chemiluminescence mode (with no excitation source).
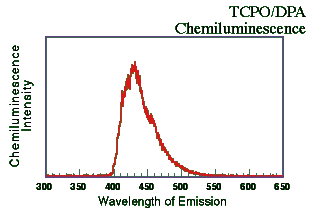
The slit and PMT voltage were adjusted to allow for the detection of a strong signal without overloading the detector. The components were mixed and the emission spectrum scanned immediately (1200 nm/min). As the Figure below shows, the emission was centered around 425 nm. This is, of course, similar to DPA's "normal" fluorescent emission.
Movie of TCPO + H2O2 + Diphenylanthracene Chemiluminescence Reaction
The movie included here involves that same solution, TCPO and 9,10-diphenylanthracene dissolved in THF. If you look closely you may be able to see the milky consistency of the slightly yellow, initial mixture--shown under fluorescent lights, before hydrogen peroxide was added. Without a mixed solvent system, the solubility of each of these components is relatively low and so the solution is basically saturated with each of these reagents (but still in the low millimolar concentration range).
In the dark, a solution of ~0.3% H2O2 in THF was added dropwise to approximately 8 mL of the fuel + fluorophore in THF (~25oC) in an open-topped vial. The reaction(s) immediately produces light from the excited fluorophore. The emission is relatively short lived but since H2O2 is apparently limiting, a second and third dropwise addition of the oxidant yields additional bursts of light. If you will look carefully at the end of the movie you will see a clear--yet still yellow--solution in which all precipitates have dissolved. Also apparent to the experimenter, but undetectable in the movie, was the formation of a gas produced by the reaction; this appeared as a bubbling that could be seen while the reaction was still producing light yet which stopped as the reaction reached completion, about 30 seconds after the last (excess) H2O2 addition. This kind of gas production has been used as evidence for the production of CO2 as a product from the 1,2-dioxetandione intermediate as detailed in the figure above. Further peroxide addition does not yield more bubbling so this is not simply H2O2 decomposition. The process of filming this reaction is described below.
Luminol Chemiluminescence
Luminol is also widely used as a chemiluminescent reagent, but unlike the peroxyoxalate systems does not require an organic/mixed solvent system. The chemiluminescent emitter is a "direct descendent" of the oxidation of luminol (or an isomer like isoluminol) by an oxidant in basic aqueous solution. Probably the most useful oxidant is also hydrogen peroxide similar to peroxyoxalate chemiluminescence; however, other oxidants have been used such as perborate, permanganate (Lu and Lu, 1992), hypochlorite (Cunningham et al., 1998), and iodine (Seitz, 1981). If the fuel is luminol, the emitting species is 3-aminophthalate (see below); however, luminol-derivatized analytes allow for determination of compounds that would not normally chemiluminescence in this system and presumably have slightly different emitters (Edwards et al., 1995; Kawasaki et al., 1985; Lippman, 1980; Nakazone et al., 1992; Pontén et al., 1996).
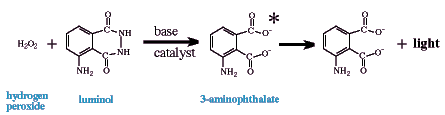
The presence of a catalyst is paramount to this chemiluminescent method as an analytical tool. Many metal cations catalyze the reaction of luminol, H2O2, and OH- in aqueous solution to increase light emission or at least to increase the speed of the oxidation to produce the emitter and therefore the onset/intensity of light production. [Some metals, however, repress chemiluminescence at different concentrations (Yuan and Shiller, 1999; see below.] This therefore can be the foundation of significantly different analytical determinations. For instance, this system can be used:
- to determine luminol itself by holding other variables constant
- to determine luminol-like derivatives similarly (Edwards et al., 1995; Kawasaki et al., 1985; Lippman, 1980; Nakazone et al., 1992; Pontén et al., 1996)
- to determine hydrogen peroxide or the progress of reactions that produce H2O2 (Yuan and Shiller, 1999; Tsukagoshi et al., 1998)
- to determine the concentrations of metal cations (Kyaw et al., 1998; O'Sullivan et al., 1995; Robards and Worsfold, 1992; TheingiKyaw et al. 1999)
- or to determine analytes that effect the concentration of metal catalysts.
This last is particularly powerful feature of this system because many compounds complex metallic cations and thereby make themselves "known." Amino acids (Koerner and Nieman, 1987), fructose and tagatose (Valeri et al., 1997), glycerol (Robards and Worsfold, 1992), thiols (Sano and Nakamura, 1998), and serum albumin (Tie et. al., 1995) among many others have been determined using luminol chemiluminescence.
Most recently, Yuan and Shiller (1999) report a subnanomolar detection limit for H2O2using luminol chemiluminescence. Their method, which was used to determine hydrogen peroxide content in sea water, was based on the cobalt(II) catalytic oxidation of luminol. While Co is the most sensitive luminol metal catalyst, it is also present in sea water at very low concentrations. The pH of the luminol solution used in this work was 10.15, and interferences from seven different metals were investigated. Interestingly some metals interfered positively and some negatively, and Fe(III) interfered positively at one concentration and negatively at another. Finally, very low concentrations of iron(II) showed a significant positive interference in determination of H2O2, but the authors used the relatively short half life of Fe(II) in marine water as a means of eliminating Fe(II) interference in the determination of hydrogen peroxide in their analysis by storing samples for over 1 hr before analysis.
Light emission from 3-APA
Approximately 15 ml of a solution containing luminol, copper catalyst, and pH controllers were placed in a glass vial at ~25oC (1 x 10-3 M luminol; 0.05 M sodium carbonate; 0.3 M sodium bicarbonate; 5 x 10-3 M ammonium carbonate; 1.5 x 10-3 M Cu(II) added as sulfate salt). An aqueous solution of approximately 0.25% H2O2 was added dropwise.
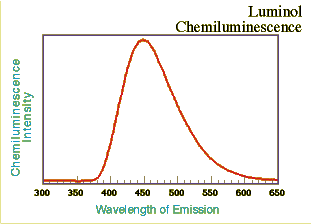
The emission spectrum was taken as before using a fluorescence spectrometer with the excitation source off. The light intensity-time decay data were taken immediately after mixing the reagents and for 60 seconds. The lambda max is at approximately 445 nm, slightly longer wavelength than the TCPO/DPA system described above. Online presentations of the light intensity-time decay aspects of the luminol reaction with hydrogen peroxide and differing concentrations of Cu(II) as catalyst are also available elsewhere (Iwata and Locker, 1998); however, with this reagent mixture the onset of emission was almost instantaneous and reached a maximum within a few seconds.
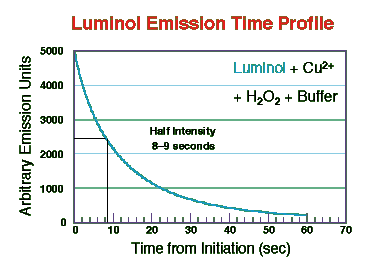
As the figure shows the light intensity decayed to approximately 50% of maximum at about 8 seconds. Iwata and Locker found that both the initial intensity and rate of decay in this kind of system was dependent on Cu(II) content. In the TCPO system described above, Orosz et al. (1996) reported that decay rate, rise constant, maximal light intensity, and quantum efficiency depended on hydrogen peroxide concentration. These authors present a comprehensive review of efforts to model the optimization of reagent flow rates and concentrations on HPLC detector responses with the TCPO reaction(s).
Movie of Luminol Chemiluminescence
The luminol reaction described above was carried out by placing approximately 15 mL of a solution containing the fuel (luminol), Cu2+ (1.5 x 10-3 M as the sulfate), and buffers detailed above in a open-topped glass vial (~25oC). The initial solution is visible at the movie's beginning as light blue in color under the laboratory's fluorescent light due to aqueous copper cations. In the dark, aqueous hydrogen peroxide (~0.3%) was added dropwise four times (small 1 or 2 mL squirts is probably a better description). The light emission is also, as before with TCPO, almost simultaneous upon mixing. The light produced appears white/blue and, as in the TCPO/DPA movie, since fuel is initially in excess, multiple injections of the limiting H2O2 reagent are necessary to take the reaction nearer to completion. Finally, after the fourth addition, the mixture was allowed to decay undisturbed and the light intensity drops off rather quickly (see the time decay data above). Approximately 80 seconds after the initial mixing began, the overhead fluorescent light were turned on and the final frame shows that solution. The light blue solution then appear green with a finely dispersed, black precipitate.
Table 1: Analytically Useful Chemiluminescent Emitters
| Chemiluminescent Emitter |
Lambda Max (Wavelength Region) |
Reference |
|---|---|---|
| 3-Aminophthalate (from luminol) |
425 nm | White and Roswell. In Burr, Ed., Chemi- and Bioluminescence, Marcel Dekker, New York, 1985, p. 215. |
| CH3Se | 750-825 nm | Glinski et al., J.A.C.S.. 108, 531 (1986). |
| CN | 383-388 nm | Sutton et al., Anal. Chem.. 51, 1399 (1979). |
| HCF | 475-750 nm | Glinski et al., J. Photochem.. 37, 217 (1987). |
| HCHO | 350-500 nm | Finlayson et al., J.A.C.S..96, 5356 (1974). |
| HF | 670-700 nm | Glinski et al., J. Photochem.. 37, 217 (1987). |
| HSO | 360-380 nm | Toby, Chem. Rev.. 84, 277, (1984). |
| IF | 450-800 nm | Getty and Birks, Anal. Lett.. 12, 469, (1979) |
| N-methylacridone (from lucigene) |
420-500 nm | Totter, Photochem. Photobiol. 22, 203 (1975). |
| Na | 589 | Gough et al., J. Chromatogr.. 137, 293 (1977). |
| NO2 | 1200 nm | Greaves and Gavin, Chem. Physics..30, 348, (1959). |
| OH | 306 nm | Toby, Chem. Rev.. 84, 277, (1984). |
| Oxyluciferin (from luciferin) |
562 nm | Seitz, Crc Crit. Rev. Anal. Chem.. 13, 1 (1981). |
| Ru(bpy)3 | 600 nm | Rubinstein et al., Anal. Chem.. 55, 1580 (1983). |
| S2 | 275-425 nm | Spurlin and Yeung, Anal. Chem..54, 318 (1982). |
| SF2 | 550-875 nm | Glinski and Taylor, Chem. Phys. Lett..155, 511 (1989). |
| SO2 | 260-480 nm | Kenner and Ogryzlo, In Burr, Ed., Chemi- and Bioluminescence, Marcel Dekker, New York, 1985, p. 139. |
Internal Links
- Luminol
Outside Links
- http://www.shsu.edu/~chm_tgc/JPPdir/JPP1999
- Gas Phase Chemiluminescence
- Solution Phase Chemiluminescence
- electrochemiluminescence
References
- Alverez, F. J.; Parekh, N. J.; Matuszewski, B.; Givens, R. S.; Higuchi, T.; Schowen, R. L. J. Am. Chem. Soc. 1986, 108, 6435-6437.
- Chokshi, H. P.; Barbush, M.; Carlson, R. G.; Givens, R. S.; Kuwana, T.; Schowen, R. L. Biomed. Chrom. 1990, 4(3), 96-99.
- Cunningham, C.; Tipton, K. F.; Dixon, H. B. F. Biochem. J. 1998, 330, 939-945.
- de Jong, G. J.; Lammers, N.; Spruit, F. J.; Brinkman, U. A. T.; Frei, R. W. Chromatographia 1984, 18(3), 129-133.
- de Jong, G. J.; Lammers, N.; Spruit, F. J.; Frei, R. W.; Brinkman, U. A. T. J. Chrom. 1986, 353, 249-257.
- Edwards, R.; Townshend, A.; Stoddart, B. Analyst 1995, 120, 117-20.
- Givens, R. S.; Schowen, R. L., The Peroxyoxalate Chemiluminescence Reaction, In Chemiluminescence and Photochemical Reaction Detection in Chromatography, J. W. Birks, Ed.; VCH: New York, 1989; pp 125-147.
- Grayeski, M. L.; Weber, A. J. Anal. Lett. 1984, 17(A13), 1539-1552.
- Iwata, N.; Locker, J. R. Final Report Summer Research: July 30, 1998; Department of Chemistry, Washington College, Chestertown, MD, USA.
- Jennings, R. N.; Capomacchia, A. C. Anal. Chim. Acta 1988, 205, 207-213.
- Kawasaki, T.; Meada, M.; Tsuji, A. J. Chromatogr. 1985, 328, 121-126.
- Kobayashi, S.; Imai, K. Anal. Chem. 1980, 52, 1548-1549.
- Koerner, P. J.; Nieman, T. A. Mikrochim. Acta 1987, II, 79-90.
- Kwakman, P. J. M.; Brinkman, U. A. T. Anal. Chim. Acta 1992, 266, 175-192.
- Kyaw, T.; Fujiwara, T.; Inoue, H.; Okamoto, Y.; Kumamaru, T. Anal. Sci. 1998, 14, 203-207.
- Lippman R. D. Anal. Chim. Acta 1980, 116, 181-184.
- Lu, X.; Lu, M. Fenxi Ceshi Tongbao, 1992, 11, 41-43.
- Milofsky, R. E.; Birks, J. W. J. Am. Chem. Soc. 1991, 113(26), 9715-9723.
- Nakazono, M.; Nohta, H.; Sasamoto, K.; Ohkura, K. Anal. Sci. 1992, 8 779-784.
- Nieman, T. Detection Based on Solution-Phase Chemiluminescence Systems, In Chemiluminescence and Photochemical Reaction Detection in Chromatography, J. W. Birks, Ed.; VCH: New York, 1989; pp 99-123.
- Orlovic, M.; Schowen, R. L.; Givens, R. S.; Alvarez, F.; Matuszewski, B.; Parekh, N. J. Org. Chem. 1989, 54, 3606-3610.
- Orosz, G. Tetrahedron, 1989, 45(11), 3493-3506.
- Orosz, G.; Givens, R. S.; Schowen, R. L.; Crit. Rev. Anal. Chem. 1996, 26(1), 1-27.
- O'Sullivan, D. W.; Hanson, Jr. A. K.; Kester, D. R. Mar. Chem. 1995, 49, 65-77.
- Pontén, E.; Appelblad, P.; Stigbrand, M.; Irgum, K.; Nakashima, K., Fresenius J. Anal. Chem. 1996, 356, 84-89.
- Pontén, E.; Glad, B.; Stigbrand, M.; Sjögren, A.; Irgum, K. Anal. Chim. Acta 1996, 320, 87-97.
- Robards, K.; Worsfold, P. Anal. Chim. Acta 1992, 266, 147-173.
- Sano, A.; Nakamura, H. Anal. Sci. 1998, 14, 731-735.
- Seitz, W. R. CRC Crit. Rev. Anal. Chem. 1981, 13, 1-58.
- Stigbrand, M.; Pontén, E.; Irgum, K. Anal. Chem. 1994, 66, 1766-1770.
- Sugiura, M.; Kanda, S.; Imai, K. Biomed. Chrom. 1993, 7(3), 149-154.
- TheingiKyaw; Kumooka, S.; Okamoto, Y.; Fujiwara, T.; Kumamaru, T. Anal. Sci. 1999, 15, 293-297.
- Tie, J.-K.; Chang, W.-B.; Ci, Y.-X. , Anal. Chim. Acta, 1995, 300(1-3), 215-220.
- Tsukagoshi, K.; Sumiyama, M.; Nakajima, R.; Nakayama, M.; Maeda, M.; Anal. Sci. 14, 409-412.
- Valeri, F.; Boess, F.; Wolf, A.; Goldlin, C.; Boelsterli, U. A. FreeRad. Biol. Med. 1997, 22, 257-268.
- Yuan, J.; Shiller, A. M. Anal. Chem. 1999, 71, 1975-1980.